
染色质和细胞核的形成在遗传物质DNA参与的生命进程中的重要性研究已经获得显著的进展 [1, 2] 。染色质本质是DNA分子缠绕在组蛋白八聚体,并压缩到一个特定的程度。关于染色质,其广义的定义还包括其他染色质相关蛋白。染色质结构的高度压缩形式、异染色质、松散压缩形式都被称作常染色质。该结构是动态的;染色质结构的改变可以影响细胞核中DNA参与的进程:如转录、复制、修复和重组 [3-6] 。
染色质是一个动态结构,负责组织基因组信息并且综合外部和内部信号,并最终确定在不同细胞类型,不同的发育时间节点,响应于不同的刺激时,哪些基因被表达,哪些不表达 [1, 7, 8] 。这些基因组中的变化并没有显示DNA序列的变化,这种变化被定义为“表观遗传”变化。染色质表观遗传变化可由以下情况引起:非正常组蛋白(组蛋白变体)的添加,ATP依赖的染色质重塑导致的染色质结构改变,组蛋白尾部化学标记的添加(组蛋白修饰)和DNA碱基上甲基基团的添加(DNA甲基化) [9] 。
染色质相关蛋白质可以写(通过染色质修饰过程的招募)和读取特定染色质结构和功能的效应子。转录因子、DNA修复蛋白、和其他相关蛋白与染色质的作用都是由染色质结构决定的。因此,我们关于染色质结构和在细胞核生命进程中作用的研究,得益于研究各种染色质结合蛋白和表观遗传标记的位置 [9, 10] 。
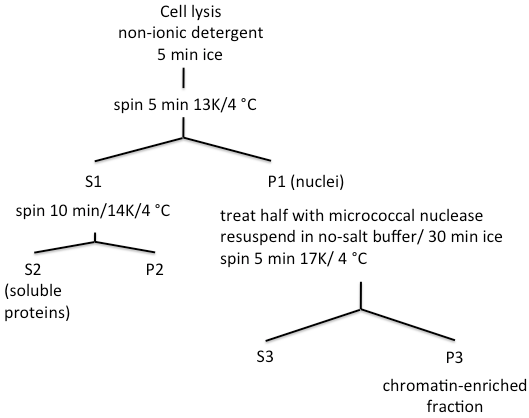
确定某个蛋白与染色质相关性的研究可采用“是或否”的问答形式:在给定的实验条件下被该蛋白质是不是染色质相关蛋白;或者确定该蛋白在基因组上的具体位置。此外,某个特定蛋白或组蛋白修饰可在全基因组上进行定位。
分析某个蛋白与染色质的相关性需要一个简单的、小型的生物化学组分分级实验。可以通过免疫印迹检测某一特定或系列蛋白的存在或缺失进行合理的组分分级。获得的信息不考虑蛋白的定位,只说明该蛋白是否与染色质相关。该方法简单,不需要大量的材料,因此,在许多需要获得充分材料可能是一个障碍的情况下相当有用。它可用于检测瞬时关联和在特定条件下的关联。尤其适用于检测在细胞特定时间节点的关联蛋白。该方法首先是用来研究酵母细胞 [11, 12] ,改进后用于研究哺乳动物细胞,后来适用于任何细胞类型 [13] 。需要注意的是,分级最后一步的离心沉淀物(P3,图一)包括与染色质结合和与细胞核基质(nuclear matrix)结合的不可溶蛋白。为区分这两者,一半的P1用0.2 U微球菌核酸酶处理,是染色质结合蛋白而不是核基质结合蛋白,变成可溶蛋白进入S3组分。用已知的关联蛋白作为对照。
| 实验 | 分辨率 | 使用难易度 | 抗体 |
|---|---|---|---|
| 生物化学 组分分级 | 低/ 非特异性地位 | 容易/ 无需优化 | 适用于免疫印迹实验(Western blotting) |
| 非变性免疫沉淀实验 | 高/ 特异性定位(和DNA直接 结合的蛋白) | 需要优化 | 适用于免疫沉淀(IP) |
| 交联免疫沉淀实验/ ChIP | 高/ 特异性定位(适用于染色质相关蛋白) | 可能需要优化 | 适用于 ChIP。 免疫沉淀的抗体可能不适用 |
ChIP实验被用来鉴定染色质相关蛋白的定位和/或它们的翻译后修饰状态。这种方法依赖于特异识别目的蛋白或修饰蛋白(例如组蛋白H3 Lys9甲基化)的抗体进行免疫沉淀和分析免疫共沉淀DNA。早期实验方法依赖于使用温和的裂解条件,以保护蛋白质--DNA相互作用,但这种方法只适用于和DNA直接结合的蛋白。甲醛交联方法的使用 [14] 使得这样的分析可以扩展到与染色质关联的几乎任何蛋白。
使用直接和特定DNA结合蛋白结合的抗体从细胞中分离蛋白质--DNA复合物依赖于抽提和免疫沉淀的条件,尤其是在该条件下怎样使蛋白可溶并保持蛋白质-DNA的结合。有几种方法已被成功使用,但是要注意到这一点,要根据蛋白质-DNA复合物所需的条件来调整实验条件 [15-17] 。
该方法本质来说是利用低渗透压裂解细胞,分离细胞核,在低盐条件下使用核酸酶(DNaseI或微球菌核酸酶—Mnase)溶解染色质,接着使用抗体进行免疫沉淀识别目标蛋白。使用多肽可以从免疫复合物中最先洗下蛋白质-DNA复合物,这可以减少在更严格的洗脱下来的,与DNA非特异性结合的蛋白污染。提取的DNA可以克隆用于进一步分析、测序或用于探针阵列分析。
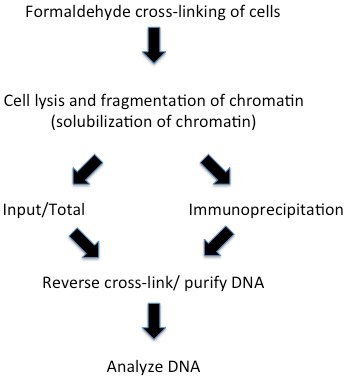
这已成为研究染色质中动态蛋白质--DNA的强有力方法 [18] 。甲醛交联的染色质免疫沉淀的实验步骤见图二。甲醛交联使我们能够检测到可能不直接结合DNA的蛋白质--染色质的结合。这种交联方法产生蛋白质-蛋白质、蛋白质-DNA和蛋白质-RNA交联,因此适合于染色质不同成分以及瞬时关联的分析。这也有效地被用于分析染色质翻译后修饰的存在与否。这种方法最初在果蝇体系中是由Varshavski及其同事开发的 [14, 19] ,由Paro 修正的 [20] 由两个酵母小组广泛使用和修正的 [21, 22] 。图二该技术实验步骤适用于所有的ChIP实验,由于研究系统的不同或研究小组的偏好,在实验细节上略有不同。此外,在新研究系统的第一次实验需要优化实验步骤。在下文我们将讨论每一个步骤的一些细节,可能的修改和不同的版本。
有许多种可用于交联反应的化学物质,但是甲醛具有多个优点而被广泛使用。甲醛可溶于水,在大幅度的条件下(缓冲液,温度等)具有活性。最重要的是,它能轻易地穿透生物膜,因此可以在完整细胞中进行交联--这降低了在制备细胞和核提取物时产生复合物重定位的可能性。甲醛可直接加入到细胞生长的培养基中,或任何其他实验材料(例如果蝇胚胎,解剖卵巢等),使其终浓度为1%即可。固定取决于时间和温度,温度越低固定速度越慢。典型的条件是在室温下时不时搅拌15分钟,但时间可以调整为在5分钟至几个小时之间。当希望快速固定时,固定终止是通过加入终浓度为0.125M的甘氨酸。甲醛处理的持续时间由给定的起始材料和目标蛋白的实验情况决定,并且要考虑可溶染色质复原时间以保证不导致目标蛋白免疫学活性的丧失,例如抗原表位被屏蔽、隐藏,以至于不能进行长时间固定。对于那些难以检测的蛋白来说,时间越短,温度越低,甚至甲醛浓度更低可能可以缓解该问题 [23] 。
| 步骤 | 优化项 | 参数改变 |
|---|---|---|
| 交联/甲醛 | 通常不需要优化 |
|
| 细胞裂解 | 通常不需要优化 | 裂解缓冲液选择:SDS-LB; FA-LB;或者其他。 |
| 染色质组分分级 | 依据需要优化 |
注意: 组分分级因裂解缓冲液不同而改变;使用特定的裂解缓冲液优化组分分级。 |
| 免疫沉淀 | 依据需要优化 | 抗体选择;抗体孵育时间: 2h-过夜 注意: IP的条件依赖于裂解缓冲液的选择。用于检测表观遗传修饰的抗体需要专门测试!!!!! |
| 解交联/ DNA纯化 | 不需要优化 | 解交联步骤按标准来;纯化: CsCl或者苯酚:氯仿提取;第二种方法更简单并且效率高。 |
| DNA分析 | 根据选择的方法进行优化 | 槽印迹(Slot blots,过时的); PCR; RT-PCR;平铺阵列(Tiling Arrays);二代测序。见表三。 |
裂解细胞和获得可溶染色质紧密关联的。依据不同的实验材料对应需要一些额外的实验步骤(例如,酵母 - 机械破损,果蝇胚胎 –去除卵壳) [21, 22, 24-26] 。在培养基和大多数其它物质中生长的细胞需要适当的裂解液重悬细胞和超声处理。甲醛处理过的细胞非常难裂解,因此超声处理这一步不仅需要更有效的裂解并且还需要染色质组分分级。作为超声处理的替代,交联的细胞核可以用微球菌核酸酶部分消化 [27] 。目前有两个主要在用的裂解液系统 - SDS-缓冲系统和FA缓冲液,见于文献 [21] 和 [22] 。也有其他实验步骤采用些许不同的缓冲液,但是这两种是使用最广的,而且在多个系统中被证明可以很好地使用。对于其他缓冲液,SDS缓冲具有许多优点 – 是超声处理最有效的裂解缓冲液;染色质片段的大小分布变化小;背景少。这个缓冲液包含1%的SDS,在免疫沉淀实验中则需要稀释至0.1%。这个缓冲液条件适用于大多数,但不是所有的抗体。如果缓冲液条件由具体实验决定(见下文),那么这个缓冲液不适合免疫沉淀实验,FA裂解液则是一种很好的替代品。对于超声来说,大多数可重复的结果是由BioRaptor完成的,染色质片段大小平均为∼300 bp [28] 。染色质的片段化是至关重要的,因为片段化程度将影响后续实验的分辨率。稍微有所不同的哺乳动物细胞裂解实验步骤是由Farnham组研发的 [29, 30].
该步骤中的关键因素是抗体选择。如果这个抗体在ChIP实验中没使用过/显示可以使用,采取预实验是非常明智的。请注意,并不是所有公司销售的“ChIP抗体”都可以用来进行ChIP实验。免疫沉淀实验中效果良好的抗体,可能在ChIP实验中并不好用。一些抗体在0.1%SDS的环境下不能很好地发挥功能,而且免疫沉淀实验需要在FA裂解缓冲液的条件下进行。单克隆抗体和合成小肽免疫的抗体可能可以或不可以很好地发挥功能,因为在交联后抗原表位可能被屏蔽了;另一方面,这些抗体具有较高的特异性。一般的多克隆抗体,或者两种或多种单克隆抗体的混合物也是可以尝试的。一旦实验确定该抗体可在交联的条件下进行免疫沉淀,则需要测试在这些条件下的抗体特异性。这可以通过在实际ChIP实验中引入阴性对照(免疫沉淀 RNAi处理的细胞,突变细胞,等等)来实现。如果能测定全基因组,那么结果就是特别真实可靠的。
表观遗传修饰研究注意事项:用于识别组蛋白翻译后修饰的抗体需要仔细检查和验证,这些抗体参与的许多交叉反应会产生假阳性,详见文献 [31] 。为解决这些问题,最近获得了一大批抗体性质信息 [32] 。对于研究人员来说,抗体验证数据库(antibody validation databases)是一个很好的资源 [33] ,他们自己也被鼓励向该数据库提交他们的抗体测试结果。
为了纯化DNA,解交联是必要的。该过程是通过将样品在65℃加热6-12h来实现的。解交联后,将样品用RNase和蛋白酶K(或链霉蛋白酶)处理,接着用苯酚:氯仿抽提DNA。早期 [20] 是采用氯化铯梯度离心纯化DNA。然而,结果表明,两个成功使用苯酚:氯仿抽提的例子说明,该方法在较低和较高真核生物中都适用 [21-23, 29, 34] 。
共沉淀DNA的分析具有几种不同方法。
通过使用序列特异性的引物进行PCR,以检测免疫沉淀实验中特定DNA序列的存在与否的研究方法,只能确定一小部分的染色体定位。
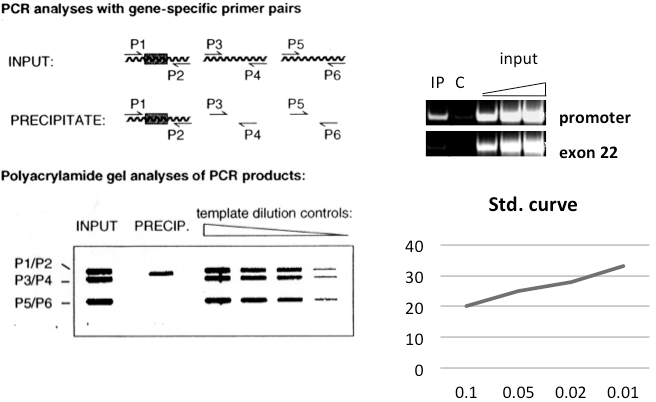
使用位点特异性引物的PCR可以表征蛋白或翻译后修饰(PTM)特定存在位置。设计用于扩增片段的引物长度在100-400 bp之间。除了研究区域的引物(图三中的引物P1/P2),还需要设计引物去扩增某一无关区域以作阴性对照(图三中的引物P3/P4和P5/P6)。多重PCR反应产物可以在丙烯酰胺凝胶上进行分离 [20, 21, 34] ,但对于普通PCR来说也有一些限制:1)片段必须以相似的扩增效率扩增;2)PCR反应必须停止在扩增的线性范围内;3)通常一个反应中不能多于3对引物。为确保PCR是在指数期,input的量需要分析研究。IP中,只有P1/P2的扩增表示特异性结合(表示的是研究区域),而input样品的所有区域都被扩增则表明是在基因组平均分布(图三,INPUT条带)。一个额外的必要对照是用非特异性抗体进行IP实验(参见IP vs. C,图三),或用目标蛋白缺失的细胞提取物进行IP实验。目标序列应该只有IP组中,而不是在对照组中(或最低限度)得到扩增。实时PCR是一种更加方便和定量的方法来分析DNA。不同片段的扩增效率不太重要,并且几乎相同大小的片段都可以被扩增。DNA Input量被用于构建每对引物的标准曲线,而IP结果则以input值的百分数表示。检测特定区域的富集度的一个有用的方法是:用该区域和非特异性区域富集度的比值进行表示 [28] 。实验设计对照是非常重要的一点,因为灵敏的PCR反应容易产生假阳性结果。相反,对于ChIP实验来说,当有其他的阳性结果时,阴性对照才有意义。
通过测序多个基因组,全基因组的整体定位研究已经成为可能。之前获得的人类全基因组序列大规模定位分析结果被用于制作CpG岛微矩阵芯片 [30, 35] 。目前,全基因组平铺阵列(tiling arrays)适用于多种生命体 [36, 37] ,但是有些生产商更倾向于用二代测序替代平铺阵列(tiling arrays)。通常,ChIP-芯片实验的第一步是扩增并同时标记IP DNA和总DNA [38] 。扩增或者由连接介导的PCR执行 [39-41] ,或者由T7聚合酶介导的体外转录执行 [42] 。标记方法见文献 [43] 。
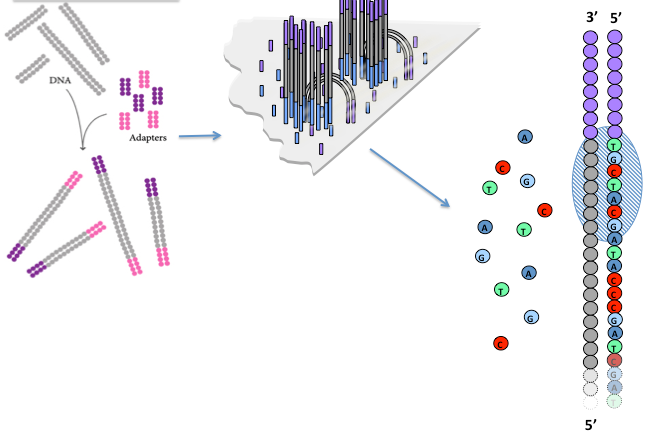
研究全基因组关联蛋白的费用越来越可接受,越来越方便,所以越来越多的研究小组研究这一领域 [44] 。有几个二代测序的不同平台,他们都提供数百万个DNA分子的实时和同时测序。图四所示的Illumina公司Solexa技术,首次被Barski和其同事使用 [45-47] 。用于测序的DNA片段两端连接上特定接头(adapter),这可以将他们固定到固体表面。然后,每个分子被放大 - 每1m集群有1000分子,每个实验有4千万个集群。每一个集群都进行合成测序。这项技术可用于多重ChIP实验,也可给不同IP实验中的DNA贴上不同的条形码,可以一次测序多种实验结果 [48] 。
类似的技术是ABI公司以前的SOLiD平台 [49, 50] ,这现在是由Life Technologies/Thermo提供 [51] ,他们公司还有另一个平台—离子流(Ion Torrent)。 二代测序技术变得更快,更便宜,更准确。因此,这项技术为我们理解染色质功能和动力学提供了前所未有的机会。然而,与其他任何实验方法类似,它也有限制因素。有关于全基因组定位研究方法的比较和保真度问题见文献 [52-55] 。
| 方法 | 优化项 | 灵敏度/保真度 |
|---|---|---|
| 普通PCR | 引物选择;扩增子长度,循环数(确保 线性范围扩增);非特异性序列选择 | 灵敏度不是最好(需要限制循环数),可以通过使用放射性核苷酸进行改善;有合适的对照时保真度高。 |
| 实时PCR | 与普通PCR一样(循环数); SYBRGreen荧光值依赖于扩增子的长度;设计合适长度的扩增子;确定能被很好检测的最短的扩增子的长度(依赖于仪器计算,通常在150 bp);每一对引物都需要作标准曲线 | 比普通PCR灵敏度高;使用标准曲线和合适的对照时具有高保真度;所有方法中保真度最高的 |
| 平铺阵列(Tiling Arrays ,全基因组) | 起始实验材料数量;扩增方法和DNA样品标记物 | 灵敏度依赖于矩阵的类型;灵敏度和特异性依赖于数据分析的算法;整体的灵敏度和保真度比二代测序低 |
| 二代测序 (全基因组) | 起始实验材料数量,PCR扩增; 测序深度;input和IP 样品测序深度相同 (input深度可以更深);单末端 vs. 双末端测序;测序平台选择;数据分析算法选择 | 可能比矩阵方法具有更高的分辨率和基因组覆盖度;更大的动态范围;分辨率和覆盖度依赖于测序深度;偏差由DNA碱基组成和染色质状态引入;灵敏度和特异性依赖于数据分析算法 |
- Badeaux A, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211-24 pubmed
- Ehrenhofer-Murray A. Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem. 2004;271:2335-49 pubmed
- Morrison A, Shen X. Chromatin modifications in DNA repair. Results Probl Cell Differ. 2006;41:109-25 pubmed
- Moss T, Wallrath L. Connections between epigenetic gene silencing and human disease. Mutat Res. 2007;618:163-74 pubmed
- Allis, C. D., Jenuwein, T., and Reinberg, D. Epigenetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. 2007.
- Donovan S, Harwood J, Drury L, Diffley J. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci U S A. 1997;94:5611-6 pubmed
- Liang C, Stillman B. Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev. 1997;11:3375-86 pubmed
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602-12 pubmed
- Solomon M, Varshavsky A. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc Natl Acad Sci U S A. 1985;82:6470-4 pubmed
- Grandori C, Mac J, Siebelt F, Ayer D, Eisenman R. Myc-Max heterodimers activate a DEAD box gene and interact with multiple E box-related sites in vivo. EMBO J. 1996;15:4344-57 pubmed
- Phelps D, Dressler G. Identification of novel Pax-2 binding sites by chromatin precipitation. J Biol Chem. 1996;271:7978-85 pubmed
- Gould A, Brookman J, Strutt D, White R. Targets of homeotic gene control in Drosophila. Nature. 1990;348:308-12 pubmed
- Kuo M, Allis C. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19:425-33 pubmed
- Solomon M, Larsen P, Varshavsky A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53:937-47 pubmed
- Orlando V, Paro R. Mapping Polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell. 1993;75:1187-98 pubmed
- Meluh P, Broach J. Immunological analysis of yeast chromatin. Methods Enzymol. 1999;304:414-30 pubmed
- Hecht A, Strahl-Bolsinger S, Grunstein M. Mapping DNA interaction sites of chromosomal proteins. Crosslinking studies in yeast. Methods Mol Biol. 1999;119:469-79 pubmed
- Williams J, Stewart T, Li B, Mulloy R, Dimova D, Classon M. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006;26:1170-82 pubmed
- Birch-Machin I, Gao S, Huen D, McGirr R, White R, Russell S. Genomic analysis of heat-shock factor targets in Drosophila. Genome Biol. 2005;6:R63 pubmed
- Austin R, Orr-Weaver T, Bell S. Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev. 1999;13:2639-49 pubmed
- Stevaux O, Dimova D, Ji J, Moon N, Frolov M, Dyson N. Retinoblastoma family 2 is required in vivo for the tissue-specific repression of dE2F2 target genes. Cell Cycle. 2005;4:1272-80 pubmed
- Cohen-Kaminsky S, Maouche-Chretien L, Vitelli L, Vinit M, Blanchard I, Yamamoto M, et al. Chromatin immunoselection defines a TAL-1 target gene. EMBO J. 1998;17:5151-60 pubmed
- Boyd K, Wells J, Gutman J, Bartley S, Farnham P. c-Myc target gene specificity is determined by a post-DNAbinding mechanism. Proc Natl Acad Sci U S A. 1998;95:13887-92 pubmed
- Farnham Lab ChIP protocols. 来自: farnham.genomecenter.ucdavis.edu/protocol.html
- Sarma K, Nishioka K, Reinberg D. Tips in analyzing antibodies directed against specific histone tail modifications. Methods Enzymol. 2004;376:255-69 pubmed
- Antibody validation databases. 来自: epigenome.ucsd.edu/antibodies.html
- Frolov M, Huen D, Stevaux O, Dimova D, Balczarek-Strang K, Elsdon M, et al. Functional antagonism between E2F family members. Genes Dev. 2001;15:2146-60 pubmed
- Wells J, Yan P, Cechvala M, Huang T, Farnham P. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene. 2003;22:1445-60 pubmed
- Affymetrix Tiling arrays. 来自: www.affymetrix.com/catalog/131463/AFFY/Human+Tiling+2.0R+Array+Set#1_1
- Graf S, Nielsen F, Kurtz S, Huynen M, Birney E, Stunnenberg H, et al. Optimized design and assessment of whole genome tiling arrays. Bioinformatics. 2007;23:i195-204 pubmed
- Lee T, Johnstone S, Young R. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1:729-48 pubmed
- Lieb J, Liu X, Botstein D, Brown P. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327-34 pubmed
- Ren B, Robert F, Wyrick J, Aparicio O, Jennings E, Simon I, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306-9 pubmed
- Gerton J, DeRisi J, Shroff R, Lichten M, Brown P, Petes T. Inaugural article: global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2000;97:11383-90 pubmed
- Liu C, Schreiber S, Bernstein B. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 2003;4:19 pubmed
- Pat Brown Lab protocols. 来自: brownlab.stanford.edu/Pat_Brown_Home_Page/Resources.html
- Zhao J, Grant S. Advances in whole genome sequencing technology. Curr Pharm Biotechnol. 2011;12:293-305 pubmed
- Solexa sequencing technology. 来自: www.illumina.com/systems/sequencing.html
- Barski A, Cuddapah S, Cui K, Roh T, Schones D, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823-37 pubmed
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651-7 pubmed
- Life Technologies Next-Generation sequencing Technologies. 来自: www.thermofisher.com/us/en/home/life-science/sequencing/next-generation-sequencing.html