
An overview of live cell imaging methods.
活细胞成像技术彻底革新了生物学家研究细胞、蛋白质以及众多分子之间相互作用和生理过程的方式。这项技术使得科学家们可以实时或者在一段时间内观察细胞内部结构和细胞生理过程。了解这些细胞结构和动态过程对于解释许多细胞生物学问题是至关重要的。与固定细胞的成像研究中提供的“快照”相比,对于动态变化的观察使得人们对细胞的运作过程的认识更加深入。此外,由于活细胞成像更不容易引入实验伪影,它通常能提供比固定细胞成像更加真实可靠的信息。这篇综述将回顾活细胞成像的系统构成、使用的探针、常见的方法和应用。
使用活细胞成像方法可以回答一系列广泛的的生物学问题。最热门的应用包括细胞结构组分的检测,动态过程的研究以及分子的细胞定位。细胞完整性、胞吞、胞吐、蛋白质转运、信号转导和酶活性等过程都可以被检测。此外,一些特定的成像系统也可以用来观测活体动物中的分子。一些常见的应用及其对应的显微成像方法以及名称标准缩写见表一。
| 实验需求 | 方法 | 名称缩写 |
|---|---|---|
| 观测分子在细胞内,以及细胞之间(通过缝隙连接)的运动 | 漂白后荧光恢复 | FRAP |
| 离子浓度(钙、镁)的时间或空间变化 | 离子成像 | 空 |
| 细胞内钙离子浓度的定量 | 比例法离子成像 | 空 |
| 生物传感 | 荧光共振能量转移 | FRET |
| 观测发生在细胞膜上或者附近的事件,或者对薄切片成像 | 全内反射显微成像 | TIRF |
| 对细胞进行长期成像 | 时域荧光寿命显微成像 | TD-FLIM |
| 观测某个分子随着时间的扩散情况 | 单分子示踪-荧光相关谱 | SMT-FSC |
| 单分子的精确分辨 | 光激活定位显微镜 | PALM |
为了获得可靠地数据,活细胞成像技术需要若干特征来保持的细胞的健康。其中最重要的一项就是为细胞提供一个最佳的生理环境。此时,科学家们不仅要确保细胞的存活,还要将细胞保持在一个稳定的代谢状态,使其不至于产生能够改变待观测过程的非特异性变化。这就包括维持温度和pH值稳定,并减少物理震动。
能提供最佳的成像条件的一些系统属性对于活体成像来说也是必要的。检测系统既必须对弱荧光样品足够敏感以获取图像,又需要在背景噪声或者样品自发荧光之上获得高的信号水平。为了捕捉动态过程,这个系统必须足够快。为了捕捉到非常好的细节,相机必须拥有足够的分辨率。为了精确的测量非常微小的光强变化,检测系统同时也必须具有一个较宽的动态范围。
在活细胞成像实验中的首要大事是在细胞内维持一个正常的代谢状态。在没有培养基或者适宜温度的条件下,细胞只能维持几分钟的正常活动。对于短期成像来说,维持细胞的健康就显得不那么重要了,但是对于长达几个小时或者几天的成像来说,就急需去注意那些能引起代谢功能改变的因素。细胞外环境的变量包括:pH值、湿度、氧气、气压、温度和渗透压 [1] 。细胞培养基的pH值通常由HEPES缓冲液来控制,这样一来就不需要5%的CO2来维持。湿度可以通过使用密封的培养皿或者加湿的环境来维持在98%附近。对于长期实验来说,氧气可以通过经常性的更换培养基或者在初期给与过量的培养基来维持。渗透压一般维持在300mosM左右,这可以通过密封的培养皿来防止蒸发或者加湿环境来实现。在活细胞成像实验中常用的气体条件为空气或者5%的二氧化碳。对于开放的,或者密封的以及空气可控的培养皿,一般还要使用HEPES缓冲的培养基来维持气压的稳定。维持细胞在最适温度是非常关键的,因为即使是几度的波动都会扰乱细胞生理机能 [1] 。温度通常由显微镜载物台加热器或者(和)物镜加热器来调控 [2] 。另外一种常见的维持温度的方式是使用整合在细胞培养皿之内的加热元件。但是,显微镜物镜作为一个散热器会抵消加热台的效果。物镜加热器可以是金属箔毯、铜管水套或者闭环加热器。为了维持温度,在培养皿内加入足量的培养基充当热质,也可以减少温度的波动。将显微镜整体装入一个箱子中并加热其中的空气是一种更加全面的加热方式。这种方式防止了温度梯度,并使得显微镜的各个部分都保持在同样的温度,但是这样一来却限制了研究者对显微镜的使用。
| 影响细胞健康的因素 | 短期研究 | 长期研究 |
|---|---|---|
| pH | HEPES缓冲液 | CO2培养箱 |
| 温度 | 载物台加热器 | 物镜加热器 内置加热单元的细胞培养皿 装在加热箱中的显微镜 |
| 湿度 | 开放的细胞培养皿 | 紧密密封的细胞培养皿 |
| 氧气 | 过量的培养基 | 在研究中更换培养基 过量的培养基 |
| 渗透压 | 密封的培养皿 | 培养皿内置加湿系统 |
在成像期间,培养细胞的培养皿必须得提供能使得细胞尽可能正常活动的条件,同时又得允许显微镜物镜能获得一个合适的成像窗口。对于短期成像(少于30分钟)来说,在垫片上盖一片盖玻片并用密封剂密封就能保证细胞状态良好。通常,作为这类细胞培养皿的垫片的是另外的盖玻片或者薄橡胶片。密封剂可以使用琼脂糖或者真空润滑油。在细胞和盖玻片之间必须用缓冲液充满。无论是生长培养基还是磷酸盐缓冲液(PBS)都能可以充当缓冲液。

对于长期成像来说,细胞培养皿则需要一些更加先进的特性。开放式的细胞培养皿和佩特里细菌培养皿类似,它们都直接与大气接触。这类培养皿中的细胞容易接触,因此研究者可以向其中加入药物,更换培养基或者实施显微注射。封闭式的培养皿则被密封起来以防止培养基的蒸发,并将细胞与外界环境隔离开来。这类培养皿可能还会具有一个用来加入药品的小孔。简单密封的的细胞培养皿有:底部有孔的细胞培养板或者是具有小腔的显微镜载玻片。前者可能会用盖玻片在底部密封,后者则用盖玻片在上面密封。它们使用起来很简便,但是密封的并不严密,并且也无法提供任何温度控制。这类培养皿必须与装备有培养皿加热单元的显微镜一起使用。对于那些需要对环境控制更加精确的细胞成像实验来说,则推荐使用完全密封的培养皿。这类培养皿密封紧密并能提供温度控制,因此它们能帮助细胞正常活动几个小时或者几天。对于需要严格控制细胞外环境或者长期的成像实验来说,最好的选择是集成在倒置显微镜中的一个细胞培养箱。
尽量减少照明是另外一种保持细胞健康的方法 [3] 。大多数细胞和组织并不耐受光照,而且紫外光会引起DNA损伤。光照也会加热样品,引起温度的波动。此外,我们已经知道将细胞暴露在荧光激发之下,会引起光毒性。随着荧光蛋白或者染料分子的激发产生的自由基会与周围的细胞组分相互作用。这种不利影响可以通过高效的显微镜设计来降低。显微镜的光路应该进行内部优化以减少所使用的激发光的能量。同时,它们也应该使用专门用于检测荧光发射光的检测器。在个别的实验参数中,可以通过使用能检测到信号的最低荧光探针浓度来减少照明。研究者还可以通过在培养基中避免酚红的使用来减少光照的副作用。酚红是一种常用的培养基指示剂染料,它具有很高的可见光吸收消光系数。避免它的使用能降低背景噪声并帮助防止光毒性。但是,即使实施了以上所有这些技术来减少光照,任然不能完全防止所有的由光引起的扰乱,因此在所有的实验中都必须设立对照组,以确保在成像过程中细胞功能没有受到不利影响。
分子荧光探针极大地提升了人们探索细胞结构和细胞过程的能力。它是指能够吸收某个特定波长的光并发射出更长波长的光(荧光)的化学基团。活细胞成像技术正是利用这些荧光探针,比如小分子有机染料或者量子点来特异性的标记感兴趣的分子。能与蛋白质天然或者人工连接的荧光探针都会在荧光成像中用到。关于荧光探针的物理特性的深入介绍已经超出了本文所讨论的范畴,也已经有学者做出了很好的综述 [4-6] 。荧光基团发展的趋势在于增加其亮度,减少细胞毒性并使得细胞成像获得高信噪比。使用荧光探针的另外一个要点是要确保它们在进入细胞时不会对细胞造成损伤。探针进入细胞的方法包括:将染料脂化以促进细胞对其吸收,使用合成囊泡来包裹探针 [7] ,以及使用机械的方法,例如显微注射和电穿孔。
天然发光的绿光荧光蛋白(green fluorescent protein,GFP)最早是从水母中分离得到的,它的发现对细胞成像来说是革命性的。作为对这项工作的认可,下村修(Osamu Shimomura)、 马丁·查尔菲(Martin Chalfie)和钱永健(Roger Tsien)在2008年因发现和改进了GFP被授予了诺贝尔化学奖。此后,人们开发了许多其它天然发光和人工改造的荧光蛋白(luorescent proteins,FPs) [8] 。荧光蛋白或者能自发荧光,或者选择性标记有一个荧光标签。荧光蛋白分子一般比较大,对于一些研究来说并不合适。这时,大小小于1kDa的小分子有机染料,例如荧光素和若丹明,或者大小小于10nm的,被称为量子点的无机纳米晶颗粒,就成为一种替代方案。与荧光蛋白相比,这类小分子荧光探针对于兴趣分子自然行为的影响更小。关于有机分子染料和量子点的优势和局限,Resch-Genger在 [9] 做了详细描述。与荧光蛋白相比,虽然有机分子染料和量子点有一些优势,但是却需要像抗体一样与目的分子相连接。
具有荧光标签的蛋白质在各类活细胞成像实验中应用广泛。这类蛋白质具备有机染料和量子点不具备的优势。比如,它们具有靶向定位亚细胞结构的能力,并且细胞毒性非常低。GFP是这类荧光探针中第一个被发现的成员。它已经被广泛应用于细胞成像和其他许多细胞和分子生物研究 [10, 11] 。GFP的荧光基团是由3个相邻的氨基酸:丝氨酸、酪氨酸和甘氨酸组成的。这个基团被氧激活后可发出荧光 [12] 。GFP应用广泛原因在于它的基因能在不损伤细胞的前下,在哺乳动物细胞中表达并产生一个可以发出荧光的功能蛋白。一些能增加表达量或者减少其温度敏感性的GFP衍生物在组织培养活细胞和活体成像实验中应用非常广泛 [13] 。将GFP的基因序列突变后可以得到蓝色荧光蛋白(BFP)、青色荧光蛋白(CFP)。这两种蛋白的激发和发射性质和野生型的GFP相比有一些改变 [14] 。黄色荧光蛋白和红色荧光蛋白衍生物的发射特性更不相同,这使得研究者可以在同一个细胞同时通用几种荧光蛋白探针。荧光蛋白的缺点在与他们的尺寸比较大(27Kd),以及其β桶装结构。后者是其发光所必须的。为了解决这些弊端,并发挥其能与细胞特异性结合,低毒性的特点,人们开发了荧光多肽。这种荧光标签的的尺寸更小,对目标天然行为的改变更少。最后,虽然GFP及其衍生物对于活细胞成像是非常有价值的工具,但是通常对于待研究的细胞来说它们都是外源蛋白。许多不发荧光的天然蛋白质可以通过共价、非共价或者酶方法来标记荧光染料 [15] 。
最传统的荧光探针有:荧光素、若丹明、吖啶橙、碘化丙啶(PI)、4',6-二脒基-2-苯基吲哚(DAPI)和Hoechst染料。这些染料大多由紫外光或者蓝光激发。异硫氰酸荧光素(Fluorescein isothiocyanate,FITC)是一种基于氧杂蒽的荧光染料。由于其高信噪比,它在宽场和共聚焦荧光显微镜中都广泛应用。但是,荧光素的激发强度对于pH的变化是非常敏感的,并且其激发光谱比较宽,会与其他荧光探针的激发光谱重叠。这就使得双重或者三重染料标记显得比较困难 [16] 。若丹明是另外一种小分子有机探针,随着吸收和发射的微小改变其名字也有许多变种。与荧光素相比,若丹明衍生物对于外部环境的依赖更小,因此也更适于多重标记实验 [17] 。吖啶橙是一种能插入DNA碱基对,或者与RNA和单链DNA结合的染料。它能在细胞膜上自由扩散,在溶酶体中积累 [18] 。碘化丙啶也能与DNA结合,而且还与双链RNA有亲和力,通常用于双重或三重标记试验中对细胞核的染色 [19] 。DAPI和Hoechst染料能与DNA双螺旋特异性的结合,同样也是一种非常常用的多重标记实验中的细胞核染色剂 [20, 21] 。

近年来,Molecular Probes公司一直在销售被称为Alexa Fluors的磺化若丹明衍生物(Molecular Probes公司已经将Alexa Fluor注册为商标) [22] 。与传统若丹明衍生物相比,Alexa Fluor系列染料具有更强的发射,光稳定性以及水溶性。Alexa Fluor系列染料的激发和发射谱各不相同,这可以根据其数字名来区分 [23] 。利用以上这些多种多样的荧光探针变体,多重标记实验可以完美的实现。Alexa Fluor系列染料既可以作为活性中间体(马来酰亚胺、琥珀酰亚胺脂、肼类),也可以预先与鬼笔环肽、糊精和大多数二抗等其它分子偶联 [23] 。花青染料的荧光特性和传统的荧光素染料类似,但是它们的光稳定性和水溶更强。这个家族包括Cy2, Cy3, Cy5, Cy7以及它们的衍生物。这些染料的活性染料形式或者和其它分子的偶联物都可以商业获取 [24] 。Cy5衍生物能被红光区域的光(650nm)激发,因此它们的发射就在远红区域(680nm)。由于大多数传统染料的激发光为紫外,发射为蓝光,因此,Cy5染料在三重标记实验中应用广泛。但是,正因为Cy5的激发在远红光区域,其发射光也只能由特殊的CCD相机来检测。
活细胞成像中也经常用到能够与无机离子,金属离子,硫醇,硫化物相互作用的荧光探针。这类探针被称为光敏感指示剂。与特定的目标离子结合后它们荧光特性会发生改变。许多指示剂,例如fura-2 和indo-1能与钙离子相互作用,它们可以用于测量细胞内的局部钙离子浓度,也可以用于定量检测质膜上的钙流的改变 [25] 。使用特定的指示剂也可以测量其他一些金属离子,如镁离子,钠离子,钾离子,锌离子等。细胞器本身也也可以用特异的荧光标签选择性的特异标记。常见的细胞器探针有MitoTracker和MitoFluor。这两种都是由Molecular Probes公司生产的用来标记线粒体的荧光探针 [26] 。图1展示了一个用MitoTracker染色肌肉细胞系的例子 [27] 。LysoTracker和LysoSensor探针常用来标记溶酶体,BODIPY及其衍生物常用来标记高尔基体。
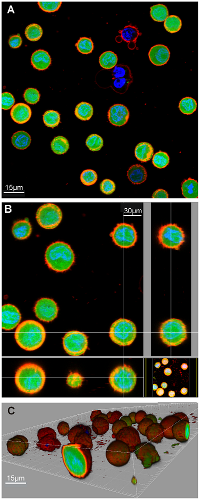
纳米尺寸的半导体晶体被称为量子点。这类荧光探针具有卓越的光稳定性,高强度的荧光,同一波长激发光可激发多种颜色,因此在宽场和共聚焦荧光显微成像中得到了普及 [28, 29] 。量子点一般由硒化镉组成,这种半导体晶体的发射能量取决于其本身的物理尺寸。只使用一种波长的光来激发不同尺寸的硒化镉量子点,就可以发射不同波长的的光。在荧光成像应用中量子点一般与抗体、蛋白质或者糖类偶联。图2展示了一个量子点成像的例子 [30] 。
| 探针类型 | 常见应用 | 优势 | 劣势 |
|---|---|---|---|
| 荧光蛋白 (FP) |
|
|
|
| 荧光多肽 |
|
|
|
| 有机分子探针 |
|
|
|
| 指示剂探针 |
|
|
|
| 量子点 |
|
|
|
显微镜是活细胞成像系统的心脏。显微成像系统既需要显微镜也需要把图像记录下来,这一般是由CCD相机来实现的。标准的光学显微镜,也被成为明视场显微镜,是利用可见光来进行成像的。尽管明视场显微镜在一些基础细胞生物学领域得到了很好的应用,但由于其相对低的对比度和分辨率,它的应用也受到了限制。既然细胞内的结构是没有颜色的,那么使用染料对细胞进行着色便能改善明视场显微镜成像的质量,但是这样做需要将样品杀死或者固定。染色也可能会引入造成样品自然状态改变的假象。暗视野显微镜能够增加未染色样品的对比度。其原理是:降低到达成像平面的非散射光,并只收集经由细胞散射的光。但是暗视野显微镜的分辨能力仍然相当低。相差显微镜比明视场或暗视场显微镜具有更好的对比度,因为它降低了直射光的强度,并创建一个四分之一波长的相位差。这使得直射光的特性发生了改变,并与衍射光发生干涉,由此产生相差图像。遗憾的是,相差显微镜只适用于较薄的样品。而在较厚的样品上会产生光晕并导致图像细节变得模糊。微分干涉相差显微镜(Differential interference contrast microscopy,DIC)比相差显微镜具有更好的分辨率。它被用于包括细胞迁移研究(伤口愈合)在内的活细胞成像试验中。微分干涉相差显微镜需要一个偏振光源,两个偏振片,以及一个特殊的分光棱镜。这个分光棱镜能将光分成两束。这两束光在细胞折射表面(如细胞核)产生的差异会在图像上产生浮雕样的效果 [31] 。
荧光显微镜在活细胞成像中应用非常广泛,它是指各种形式的使用荧光染料对分子进行着色的显微成像技术。由于能够突出细胞环境内的感兴趣的特异结构,这项技术对细胞生物学领域,特别是活细胞成像领域极其重要。用来激发荧光染料的激发波长和发射波长是不相同的。因此,通过采集发射光我们就可以得到荧光染料所标记的特异分子或结构的图像。旨在阻止激发光到达检测器的滤光片非常重要。和明视场显微镜、暗场显微镜以及相差显微镜相比,荧光显微镜优势在于可以同时使用不同的荧光染料来检测细胞的不同组分,而且这些检测可以同时进行。
共聚焦显微镜使用汇聚的光束或者激光成像,而不是对样品进行宽场光成像 [32] 。共聚焦激光扫描显微镜(Confocal laser scanning microscopy,CLSM)使用一束的聚焦的单激光束,以及一个成像针孔和检测器 [33] 。转盘共聚焦显微镜则使用多束激光以及一系列基于尼普科夫转盘原理移动的针孔,这样做显著地相提高了成像的速度 [34] 。共聚焦显微镜能实现光切片。这一过程使得系统能够获得一个样品内若干不同深度的焦点图像,然后通过点对点配准方式将这些图像重建成一个样品的3D图像。图3展示了一个三色荧光共聚焦成像以及3D图像重建的例子 [35] 。共聚焦图像是通过空间滤波来消除厚样品中非焦平面的光的。CLSM 和转盘共聚焦显微镜传具备统光学显微镜所没有的一些优势。这两项技术的空间分辨能力以及对更厚样品成像的能力都有很大提升,并能更加有效的降低背景噪声。它们也能够控制成像的深度,并对厚样品进行光学切片。多光子显微镜(Multiphoton microscopy,MPM)是一类非线性激发能量在一个薄平面激发荧光的激光扫描显微镜,它能够改善3D成像的质量 [36] 。MPM的成像深度可以达到几百个微米,对于厚切片、组织以及活动物成像非常有价值。
全内反射荧光显微镜(Total internal reflection fluorescence microscopy,TIRFM)选择性的激发样品特定的(与玻璃-水界面相邻的)区域内的荧光探针并对其成像 [37] 。当入射光在玻璃-水界面发生全反射的时候,会产生一个衰逝波,并开始指数型衰减,因此大约能穿透样品大约100nm。这项技术在对质膜的研究,例如胞吐过程的研究中应用广泛 [38] 。TIRFM也是一种用来观测单分子的非常有价值的方法 [39] 。
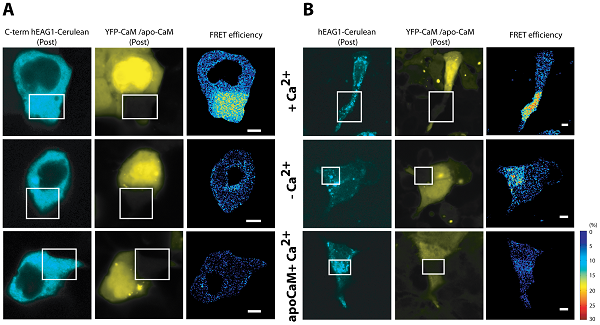
荧光共振能量转移(Forster resonance energy transfer,FRET)显微成像是一种同时使用两种荧光探针的成像技术。其中一个荧光探针的发射谱与另一个荧光探针的激发谱重叠。前者被称为FRET供体,后者被称为受体。当供体和受体分子之间的距离小于10nm的时候,对于供体探针的激发会引起能量转移导致受体的发射。在FRET成像中ECFP和EYFP通常被作为一个荧光探针对来使用。FRET现象可以用于目的分子构象改变的研究:将受体分子和供体分子设计到一个能和目的分子发生相互作用的荧光蛋白内。当目标分子被激活并且其构象产生改变时,受体和供体探针就能够靠的足够近以产生FRET信号。使用这种方法我们已经能够检测包括钙 [40] 、蛋白酶 [41] 、cAMP [42] 和激酶 [43] 在内的一系列分子。图4展示了来自 [44] 中的一个FRET成像的例子。
荧光寿命成像显微镜(Fluorescence Lifetime Imaging Microscopy,FLIM)可以观测荧光分子的处于激发态的寿命 [45] 。FILM可以依据激发寿命将FRET供体和非相互作用的供体区分开,并提高FRET的信噪比。FILM的空间分辨率很高 [46] 。它通常用于长时间的活细胞观察。最近,又发展出宽场多参数荧光寿命成像显微镜(wide-field multi-parameter FLIM,WFMP-FLIM)。这种方法采用了常规的荧光显微镜,但是对视场同时进行双波长成像,并采用一个空间敏感的光子计数器进行检测 [47] 。时域荧光寿命成像(Time domain fluorescence lifetime imaging microscopy,TD-FLIM)能够在皮秒和亚微米的分辨率下,直接检测荧光探针的荧光衰退率。
| 显微镜类型 | 技术 |
|---|---|
| 光学显微镜 |
|
| 常规显微镜 |
|
| 共聚焦荧光显微镜 |
|
| 高级荧光显微镜 |
|
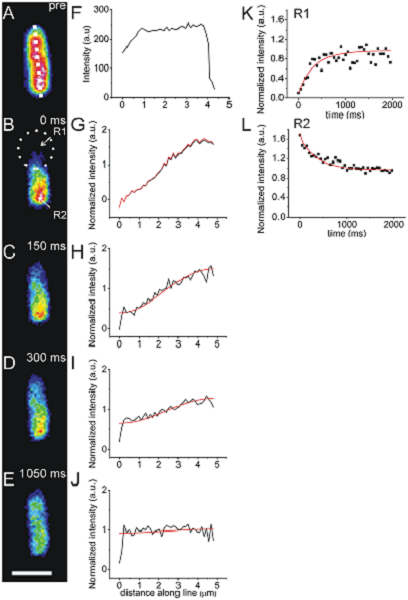
活细胞成像通常被用来观测细胞内的分子运动。目前已有多项显微成像技术能够实现这一目标。漂白后荧光恢复(Fluorescence recovery after photobleaching,FRAP)就是这样一项技术。在进行FRAP实验时,需要使用高强度的光将亚细胞区域光漂白,然后检测荧光随时间恢复的情况。荧光恢复的速率取决于荧光分子扩散或者主动运输到漂白区域的数量 [48] 。图五展示了一常规的FRAP的例子 [49] 。FRAP还可以用来研究分子通过缝隙连接的运动过程。漂白后荧光损失实验(Fluorescence loss in photobleaching,FLIP)和漂白后荧光恢复实验类似,但是漂白区域是被持续的漂白。
荧光相关谱(Fluorescence correlation spectroscopy,FSC)是一种通过测量小体积内短时间荧光强度涨落来获取目标分子扩散速率和浓度的方法 [50] 。单分子示踪技术(Single-molecule tracking,SPT)能够检测荧光标记的单分子随时间变化的扩散过程。这种方法对要求非常敏感的相机和先进的专业技术,但是它有助于人们理解诸如驱动蛋白和动力蛋白的马达蛋白的运动 [51] 。
光激活定位显微成像(Photoactivated localization microscopy,PALM )每次能随机的获得很少一部分染料分子的荧光发射图像,将这些图像组合起来就可以得到一副PLAM图像。PALM获得的图像空间分辨率非常高,可以到达20nm。PLAM需要使用能被激活并转化的荧光分子,而且由于其相对较高的背景噪声,它的应用受到了限制。但是,Matsuda等人最近发表了他们使用改进的算法来处理背景噪声并采用这种方法来成像染色质结构的工作 [52] 。
对于上述的显微镜技术来说,图像分析是非常重要的,在其它的文献中也做了全面的回顾 [53] 。图像处理的基本目标是将图像转变成有意义的信息。主要需要关注以下三个方面:降低背景噪声,增强对比度以及对信号强度进行定量。为此人们开发了许多的软件来解析图像数据,其中有不少是开源的 [54, 55] 。图像处理和数据分析的算法有很多,而不同的图像的类型和不同的研究样品需要的算法是各不相同的。在图像分析中优化图片质量和分辨率的同时,也应该注意到操作数字图像的基本准则,那就是确保图片任然能够反映其原本蕴含的原始信息 [56] 。
- Thal D, Schultz C, Botez G, Del Tredici K, Mrak R, Griffin W, et al. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer's disease-related pathology. Neuropathol Appl Neurobiol. 2005;31:270-9 pubmed
- Lowndes R, Hallett M. A versatile light microscope heating stage for biological temperatures. J Microsc. 1986;142:371-4 pubmed
- Hanson G, Hanson B. Fluorescent probes for cellular assays. Comb Chem High Throughput Screen. 2008;11:505-13 pubmed
- Miyawaki A, Sawano A, Kogure T. Lighting up cells: labelling proteins with fluorophores. Nat Cell Biol. 2003;0:S1-7 pubmed
- Fisler R, Cohen A, Ringer S, Lieberman E. Neonatal outcome after trial of labor compared with elective repeat cesarean section. Birth. 2003;30:83-8 pubmed
- Li Castri Patti L, Parodi Giusino U. [Contribution to the nosological classification of parietal dysplasias of the gallbladder]. Minerva Chir. 1967;22:344-57 pubmed
- Zimmer M. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem Rev. 2002;102:759-81 pubmed
- Heim R, Prasher D, Tsien R. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 1994;91:12501-4 pubmed
- Johnson I. Fluorescent probes for living cells. Histochem J. 1998;30:123-40 pubmed
- Wessendorf M, Brelje T. Which fluorophore is brightest? A comparison of the staining obtained using fluorescein, tetramethylrhodamine, lissamine rhodamine, Texas red, and cyanine 3.18. Histochemistry. 1992;98:81-5 pubmed
- Darzynkiewicz Z. Differential staining of DNA and RNA in intact cells and isolated cell nuclei with acridine orange. Methods Cell Biol. 1990;33:285-98 pubmed
- Arndt-Jovin D, Jovin T. Fluorescence labeling and microscopy of DNA. Methods Cell Biol. 1989;30:417-48 pubmed
- Durand R, Olive P. Cytotoxicity, Mutagenicity and DNA damage by Hoechst 33342. J Histochem Cytochem. 1982;30:111-6 pubmed
- Kubista M, Akerman B, Norden B. Characterization of interaction between DNA and 4',6-diamidino-2-phenylindole by optical spectroscopy. Biochemistry. 1987;26:4545-53 pubmed
- Panchuk-Voloshina N, Bishop-Stewart J, Bhalgat M, Millard P, Mao F, Leung W, et al. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J Histochem Cytochem. 1999;47:1179-88 pubmed
- HAINAUX H. [A REFORM OF A REFORM]. Concours Med. 1963;85:4907-11 pubmed
- Ballou B, Fisher G, Waggoner A, Farkas D, Reiland J, Jaffe R, et al. Tumor labeling in vivo using cyanine-conjugated monoclonal antibodies. Cancer Immunol Immunother. 1995;41:257-63 pubmed
- Helm P, Patwardhan A, Manders E. A study of the precision of confocal, ratiometric, Fura-2-based [Ca2+] measurements. Cell Calcium. 1997;22:287-98 pubmed
- Keij J, Bell-Prince C, Steinkamp J. Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry. 2000;39:203-10 pubmed
- Jaiswal J, Mattoussi H, Mauro J, Simon S. Long-term multiple color imaging of live cells using quantum dot bioconjugates. Nat Biotechnol. 2003;21:47-51 pubmed
- Gao X, Yang L, Petros J, Marshall F, Simons J, Nie S. In vivo molecular and cellular imaging with quantum dots. Curr Opin Biotechnol. 2005;16:63-72 pubmed
- Allen R, David G, Nomarski G. The zeiss-Nomarski differential interference equipment for transmitted-light microscopy. Z Wiss Mikrosk. 1969;69:193-221 pubmed
- NIJLAND M. [Magnoflorine in Caltha palustris L]. Pharm Weekbl. 1963;98:261-3 pubmed
- Paddock S. Confocal laser scanning microscopy. Biotechniques. 1999;27:992-6, 998-1002, 1004 pubmed
- Denk W, Strickler J, Webb W. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73-6 pubmed
- Reck-Peterson S, Derr N, Stuurman N. Imaging single molecules using total internal reflection fluorescence microscopy (TIRFM). Cold Spring Harb Protoc. 2010;2010:pdb.top73 pubmed
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137-41 pubmed
- Mahajan N, Harrison-Shostak D, Michaux J, Herman B. Novel mutant green fluorescent protein protease substrates reveal the activation of specific caspases during apoptosis. Chem Biol. 1999;6:401-9 pubmed
- DiPilato L, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513-8 pubmed
- Ting A, Kain K, Klemke R, Tsien R. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci U S A. 2001;98:15003-8 pubmed
- Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16:19-27 pubmed
- Pelet S, Previte M, So P. Comparing the quantification of Forster resonance energy transfer measurement accuracies based on intensity, spectral, and lifetime imaging. J Biomed Opt. 2006;11:34017 pubmed
- Reits E, Neefjes J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3:E145-7 pubmed
- Van Craenenbroeck E, Engelborghs Y. Fluorescence correlation spectroscopy: molecular recognition at the single molecule level. J Mol Recognit. 2000;13:93-100 pubmed
- Kural C, Kim H, Syed S, Goshima G, Gelfand V, Selvin P. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement?. Science. 2005;308:1469-72 pubmed
- Collins T. ImageJ for microscopy. Biotechniques. 2007;43:25-30 pubmed
- Yoo T, Ackerman M, Lorensen W, Schroeder W, Chalana V, Aylward S, et al. Engineering and algorithm design for an image processing Api: a technical report on ITK--the Insight Toolkit. Stud Health Technol Inform. 2002;85:586-92 pubmed
- 来邦网
- 英文来邦