
DNA甲基化是一种影响CG双碱基的复制后化学修饰反应。在该过程中,甲基基团(CH3)以共价键的方式被修饰到CG碱基的胞嘧啶C上 [1] 。该修饰是被一种叫做DNA甲基转移酶(Dnmts,DNA methyltransferases enzymes)催化反应的,其发生的部位是CG双碱基的胞嘧啶C的碳5位置,通常称作CpG位点 [2] (图1)。CpG位点的甲基化是动态变化的,而且甲基化谱式在不同的基因组也是各不相同的 [2-4] 。CpG位点的高频率甲基化区域通常位于基因的启动子区,被称作”CpG岛’’ [5] 。由于DNA甲基化是一种酶催化的甲基基团转移到CpG双碱基上的过程,因此这就需要有甲基供体化合物来提供甲基基团。甲基基团一般来源于饮食,常见的有叶酸、甜菜碱(betaine)和维生素B12。这些甲基供体化合物最终可以影响甲硫氨酸和S-腺苷甲硫氨酸(SAM)的代谢 [6] ],其中SAM是某些甲基转移酶最主要的甲基供体 [7] 。迄今为止人们已经了解了一些甲基转移酶(Dnmts),其作用各不相同但有时候会有某些重叠。Dnmt3A和Dnmt3B涉及到发育早期未甲基化DNA的甲基化谱式的建立,因此被称为从头的甲基转移酶(de novo methyltransferase) [8] 。而Dnmt1则倾向作用于半甲基化的DNA,对于维持DNA甲基化谱式至关重要 [9] 。DNA甲基化复合物机器在一些藻类、真菌、植物、无脊椎动物和脊椎动物中都有被发现 [10] 。

对DNA甲基化影响基因表达最广为人知的机制是甲基化影响了促转录因子与DNA的结合 [11] ,从而抑制了基因的表达。但是,DNA的甲基化也能促进基因的表达,这是因为DNA甲基化可以影响某些绝缘子的结合,从而允许附件增强子发挥作用 [12, 13] 。
在某些关键时期DNA甲基化谱式特别容易被重编程,这些关键时期包括受精到囊胚植入前和胚系分化的早期 [14] 。但是其他一些敏感时期也有被描述 [15] 。尽管在子宫内的表观重编程被传统认为是不可逆的,但近期一些研究表明母亲饮食介导的表观遗传学变化可以在后代的青少年时期被摄入的叶酸逆转 [16] ,虽然其中的机制还不是特别清楚。
胚系发育起始大部分表观遗传印记会被擦除 [14, 17] ,然后在性别决定的起始时期DNA甲基化谱式会被重建 [18, 19] 。除此之外,在胚系发育的起始期还会涉及其他表观遗传的变化 [20] 。该表观遗传重编程的时期代表着个体发育对外界因素的敏感时期,而且这一表观遗传谱式可以被诱导且在代际间永久传递 [21, 22] 。
近年来研究比较清楚的是DNA的甲基化和去甲基化对于发育期间的表观遗传调控是必不可少的生物学事件
对于成年个体疾病的病原学机制,研究者一般认为其发育过程中的表观遗传学进程是疾病的发源起点 [25, 26] 。近来发现一些列疾病均包含有表观遗传学元素的病因,比如过敏反应 [27] 、肝癌 [28] 、胃癌 [29] 、哮喘 [30] 、结肠癌 [31] 、前列腺癌 [32] 、HIV的潜伏 [33] 、代谢性疾病 [34, 35] 以及心脑血管疾病等 [36] 。还有些研究描述了DNA甲基化与母系影响,以及社会生物学层面的表现,如行为 [37] 、抑郁 [38] 和脑部疾病 [39] 之间的关系。因此,使用生物信息学以及一些列可靠的生物学研究方法对检测DNA甲基化显得及其重要。
在过去30年,随着表观遗传标志物挖掘技术的进步,人们才得以深刻理解表观遗传学在医学和生物学中所扮演的角色。 目前DNA甲基化的研究方法分为三种:全局甲基化(global methylation)、局部甲基化(local methylation),以及基因组尺度的甲基化(genomewide methylation)。 全局甲基化分析是第一种发展起来的分析方法,其目的主要用来确定基因组整体的DNA甲基化水平,局部的甲基化变化是不在考虑范围内的。该方法主要涉及用来源于SAM的放射性甲基供体被甲基转移酶Sss1催化修饰到DNA样品之中 [40, 41] 。还有利用CpG位点甲基化敏感的限制性内切酶的切割活性来区分DNA甲基化与否 [42, 43] 。近年来用来测量DNA全局甲基化的方法还包括通过胞嘧啶甲基化特异性抗体富集甲基化然后用荧光定量法检测; [44, 45] 还有用高效液相质谱 [46-48] 结合UV(HPLC-UV) [49] 或者串联质谱直接定量甲基化胞嘧啶(LC-MS/MS) [50] ;酶联免疫的方法(ELISA) [51] 以及使用短重复序列 [52] 或者线性原件 [53] 代表基因组的甲基化状态,然后其中的5mC可以用生物素-链亲和素(biotinstreptavidin)免疫法 [54] 、ELISA或者焦磷酸测序来检测 [55] 。全局甲基化分析方法最主要的限制在于其只能检测整体的DNA甲基化改变,而忽略局部的变化。然而,正是局部DNA甲基化的变化才是相对重要的,这是因为化合物带来的大部分效应都只体现在局部而非全局甲基化的改变。

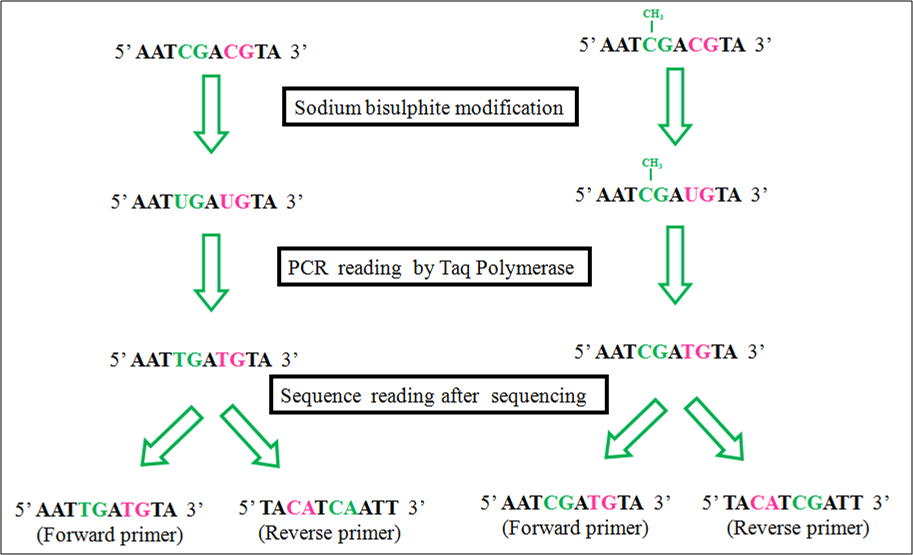
局部甲基化分析是指特异基因或基因组特定区域的甲基化谱式分析。起初利用甲基化敏感性差异的限制性酶切并结合PCR扩增 [56] 和Southern杂交 [42] 用来分析特局部甲基化状态。最常用的一对限制性内切酶是HpaII和其同列酶MspI [42, 43, 57] 。这两个酶都是识别CCGG并切割CG中间位置。但是,MspI是甲基化不敏感的,这意味着无论是否发生甲基化均可以切割CG。而HpaII,当CG发生甲基化时不能切开CG(图2)。然而,能够在CpG位点水平分析局部DNA甲基化变化的最常用方法则是亚硫酸盐测序法。这一方法最初由Frommer et al [58] 描述并经过很多研究者的优化 [59, 60] 。其原理是亚硫酸盐可以把非甲基化的胞嘧啶C转换成尿嘧啶U,但对甲基化的胞嘧啶则没有影响。然后再经过PCR扩增并测序,从而区分这种转换的差异。测序确定甲基化的C依然是C,而非甲基化的C被转变成U,经过PCR扩增变成T(图3)。其实亚硫酸盐处理后可以使用不同的方法进行检测,比如COBRA法(组合亚硫酸盐限制性分析) [61, 62] ,即PCR扩增亚硫酸盐处理的样本,再经过限制性酶切分析;直接测序法 [63] ;克隆测序 [64] ;焦磷酸测序 [65] 或质谱分析 [66] 。甲基化特异性的引物的设计程序也有现成的,如Methprimer (http://www.urogene.org/methprimer, [67] ) 。如果利用实时荧光定量PCR的方法来分析甲基化特异性的PCR产物则可以分析溶解曲线和Ct值的变化 [68, 69] 。还有一种基于PCR检测方法的变种,COLD-PCR,利用较低的变性温度来偏向性的扩增非甲基化DNA片段 [70] 。直接测序的方法可以对几个位点同时进行。但其中也存在问题,比如测序时T对C的信号强度过高导致C的信号被掩盖。而且,测序软件给出的不是定量的信号,而是经过标准化处理过的,这并不能真正反应某个CpG位点的测序峰中各自叠加T/C信号的真实比例。鉴于此,通过直接测序来测量DNA甲基化则需要通过算法或者实验来加以校正。 [71] 比如其中一种校正的算法是通过其他位置T的信号作为参考来校正T对C的信号强度过高的问题 [71] 。最近,研究者制定出一套完整的亚硫酸测序分析方法流程 [72] 。另外,亚硫酸盐处理转换之后直接单克隆测序则可以校正这一问题。这是因为直接单克隆测序代表是某单个起始DNA分子位点的状态 [60] 。但这又会带来新的问题:要检测的分子越多则需要测序的克隆也越多;如果只有少数几个克隆被测序分析又会引起较大的误差。而数字亚硫酸盐测序法是通过稀释实现单分子水平扩增的方法来模拟DNA甲基化信号,从而可以代替单克隆测序的分析方法 [73] 。尽管焦磷酸测序法 [65] 可以产生定量区分甲基化状态信号,但其缺点是每次扩增分析的序列很短,平均只有150bp左右,这限制了对CpG的分析。而亚硫酸盐处理结合质谱检测的技术 [66] 却可以极大增加分析DNA甲基化片段的长度(高达500bp)。
也有研究者开发出了基于电泳技术的甲基化DNA分离方法 [74] 。该方法要求使用变性梯度聚丙烯酰胺凝胶,十分费时费力。近年来研究者开发出了基于甲基结合蛋白(MBD)的快速显色分析法用以分析全基因组或者低丰度特异基因的甲基化状态 [75] 。首先基因组DNA被限制性内切酶消化,然后用Kelnow聚合酶和生物素标记的dNTPs(biotin-dNTPs)补平DNA的末端,之后用MBD的免疫磁珠分选,最后用链亲和素(SA)标记的HRP(辣根过氧化物酶)(SA-HRP)来定量分析甲基化DNA的量(图4)。如果要检测特定基因的甲基化,可以用biotin-dNTPs来扩增该基因,然后用SA 标记的磁珠或SA-HRP来检测。这一方法也可以和电化学方法结合使用。

第一种在基因组层面考察特定基因或者特异区域DNA甲基化的方法也是利用甲基化敏感差异的限制性内切酶分析法实现的。AIMS(Amplification of InterMethylated Sites)方法就是利用甲基化敏感的SmalI及其甲基化不敏感的同列酶PspAI酶切基因组DNA,然后连上接头进一步做全基因组水平的PCR反应 [76] 。差异性扩增代表着DNA甲基化状态的变化。类似的方法如HELP (HpaII tiny fragments Enrichment by Ligationmediated PCR),它利用的是HpaII(甲基化敏感)和其同列酶MspI(甲基化不敏感) [57] 。
在全基因组尺度解析区域DNA甲基化变化的最常用的方法之一是一种基于甲基化片段的免疫沉淀(MeDip, chromatin immuneprecipitation of methylated fragments)的方法 [77] 。这一方法是利用抗甲基化胞嘧啶的抗体(MeDIP)免疫沉淀(IP)富集甲基化的DNA片段,然后再和芯片杂交用以鉴定DNA甲基化的位点(MeDIP-chip) [77] 。这一方法被用来绘制拟南芥 [78] 、人乳腺癌 [79] 以及人的主要组织复合物(MHC) [80] 的甲基化图谱。MeDIP-chip的方法对于绘制全基因组水平的甲基化变化具有显著的优势。但是,假阳性的问题需要使用局部甲基化的分析方法进行验证。另外一种分析全基因组甲基化状态变化的方法是利用甲基化依赖的内切酶McrBC把甲基化的DNA片段去除,然后通过芯片杂交和对照比较分析哪些片段消失了 [81] 。最近,有研究者使用Infinium HumanMethylation27 Bead人甲基化芯片来分析疾病相关的DNA片段的甲基化变化 [82, 83] 。该方法是Golden Gate SNP基因分型分析方法的变种,因为目的位点SNP (C/T转换)就代表CpG甲基化的改变。
人甲基化芯片HumanMethylation27 Bead Array的升级产品Infinium HumanMethylation450 (HM450) [84] 在近年来应用的更为广泛。该芯片可以检测包括人DNA编码区、非编码区(包括miRNA启动子、5’和3’ UTR)在内的~485,000个CpG位点。还有另外一种同样来自Illumina的甲基化芯片VeraCode Methylation Array可以用以非人类DNA的分析 [85] 。得益于这些分析芯片的普及,近年来产生了大量的数据。 [86-89] 相应的,对数据的处理流程 [90] 以及基于电脑的预测方法也在持续发展之中。
MeDIP也常常和测序结合起来,由于MeDIP是先免疫沉淀甲基化的DNA片段,这样起始的测序量会精简很多 [91] 。也有另外的DNA甲基化捕捉技术,如利用甲基结合蛋白(MeCP2)结合测序的分析技术,全称是MethylCapseq 技术 [92] 。有时候研究者会首先使用甲基化不敏感的限制性内切酶MspI消化基因组从而产生包含大部分CpG的片段,这样可以使起始测序量降低到原来的1% [93, 94] ,消化的片段经过亚硫酸盐处理并测序。这一方法的被称为RRBS(Reduced Representation Bisuphite Sequencing) [94] 。比较而言,MeDIPseq, MethylCapseq 和RRBS得到的都是关于甲基化DNA的精确数据;不同的是MeDIPseq和 MethylCapseq法覆盖的基因组位置更为广泛,而RRBS对CpG含量较低的区域覆盖有限 [95] 。MethylCapseq法比RRBS法可以检测出更多的差异甲基化区域(DMR,differentially methylated regions),而RRBS比MeDIP法能检测更多的DMR [95] 。
最近有研究者比较了MethylCapseq和Illumina的HM450芯片发现这两种方法基本是互补的 [96] :HM450更加灵敏,而MethylCapseq可以覆盖更多的甲基化位点。
进年来出现了一种极具希望、不但能精准定量而且分辨率可达到CpG水平的全基因组DNA甲基化分析方法---深度测序。这一强大的技术被用来绘制拟南芥 [97] 和人 [98] 的甲基化图谱。这一方法也是利用亚硫酸盐处理然后连上接头进行全基因组测序。但是,和DNA甲基化富集再测序的方法相比比较昂贵,这也限制了该方法的应用。最近出现一种称作单分子实时测序(SMRT, singlemolecule, real time sequencing)的方法 [99] 。这一方法是把荧光标记的核苷酸掺入DNA的互补链中。碱基掺入动力学的差异可以用以区分表观遗传标记,比如甲基化、羟基化等,从而跳过了亚硫酸盐转换的步骤。以上所述的DNA甲基化分析方法的优缺点在表1内有一个简单的总结。

在过去几年,DNA甲基化研究是表观遗传学领域的大热门。在哺乳动物体内,DNA的去甲基化是有TET介导的5mC(5methylcytosine)氧化成5hmC(5-hydroxymethylcytosine,5羟甲基胞嘧啶),5fC (5-formylcytosine,5甲酰基胞嘧啶)和5caC(5-carboxylcytosine,5羧基胞嘧啶)。对此目前有两种主要的检测方法:一种是亚硫酸盐处理非依赖的,另外一种是基于亚硫酸盐处理结合甲基胞苷的转移酶处理。
亚硫酸盐处理非依赖方法检测全基因组水平的5fC。该方法主要是利用化学标记5fC,并偶联生物素用以富集,然后连上接头并用二代测序技术来鉴定5fC位点(化学标记的5fC在PCR过程中会转变成T) [100] 。
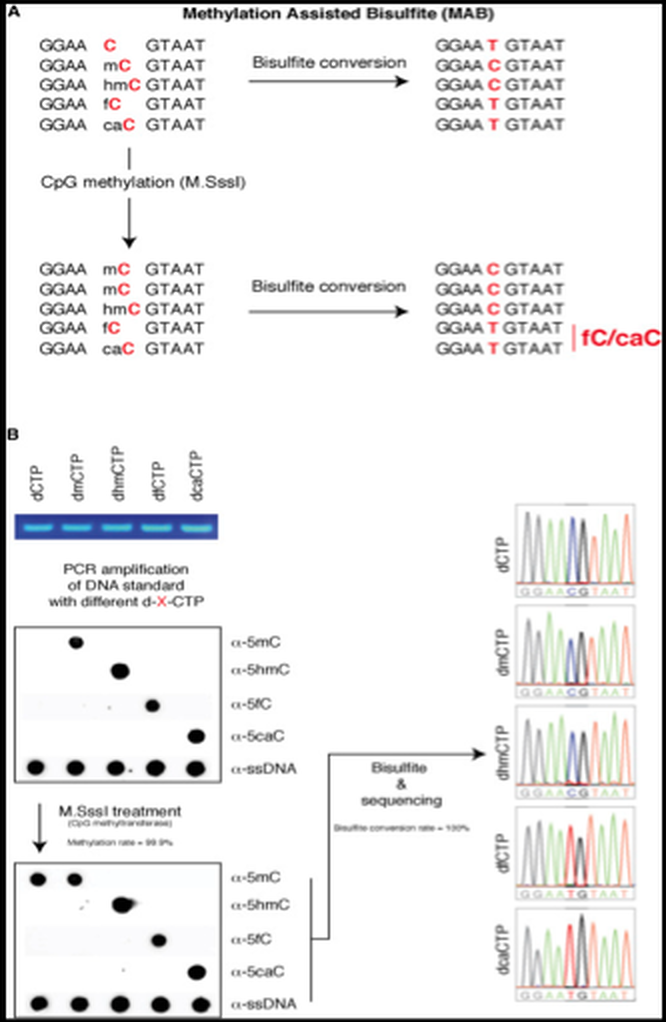
第二种方法称作甲基化辅助的亚硫酸盐测序法(MAB-Seq)。这一方法是用来在单碱基水平检测5fC和5caC。其原理是在亚硫酸盐处理之前先用基于SAM的甲基转移酶M.SssI处理DNA(这样非修饰的C就转变成5mC),然后再检测5fC 和5caC(图5, [101] )。一种改进的方法叫做RRMABseq(reducedrepresentation MABseq) [102] ,这一改进可以提高CpG区域的覆盖率,降低成本。
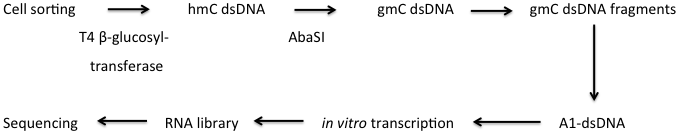
基于AbasI测序技术 [103] 是利用限制性内切酶AbaSI选择性的结合5gmC而不是5mC或者没有修饰的C,从而在结合位点的3’端产生双链DNA缺口。AbasI倾向识别的序列如图6A所示。该方法的原理是利用T4 β¬-葡萄糖基转移酶把5hmC转化成5gmC,然后经AbasI消化,连接带有3’突出端生物素修饰的DNA双链接头。然后经片段化并富集,再在另一端连上含有dT的接头,从而有利于下一步dA尾巴结合。然后经PCR扩增并测序(图6B)。
在这一方法基础上,Mooijman 等人 [104] 发展了一个在单个细胞上的全基因组水平检测5hmC的方法。在这一方法中,研究者用细胞特异的条形码序列、Illumania 5’-接头和 T7启动子代替了生物素化的接头。葡萄糖基化的DNA在体外被转录用以测序。获得的RNA被打断并整合到RNA测序文库中(图7)。
| 类别 | 方法描述 | 优点 | 缺点 | 参考文献 |
|---|---|---|---|---|
| 全局甲基化分析 | 放射性标记甲基基团 | 廉价;能全覆盖CpG位点 | 不适合用来研究局部甲基化;要放射性标记 | [40, 41] |
| 甲基化敏感差异的限制性酶切 | 廉价;非放射性;最基本的实验硬件条件 | 不适合用来研究局部甲基化;不能完全覆盖CpG位点 | [42, 43] | |
| 甲基胞嘧啶结合蛋白富集结合荧光定量 | 非放射性;能全覆盖CpG位点 | 不适合用来研究局部甲基化;要求的硬件和抗体比较贵 | [44, 45] | |
| HPLC检测 | 非放射性;能全覆盖CpG位点 | 不适合用来研究局部甲基化;要求的硬件比较贵;仪器要求非常高级的专业知识 | [46-48] | |
| 重复序列检测DNA甲基化 | 非放射性;最基本的实验硬件条件 | 不适合用来研究局部甲基化;不能完全覆盖CpG位点 | [52, 53] | |
| LC-MS/MS | 非放射性;样本需求量小 | 要求特殊仪器并需要非常高级的培训 | [50]||
| ELISA | 非放射性;最基本的实验硬件条件 | 特异性要求和抗体成本较高 | [51] | |
| 生物素-SA-HRP介导的显色反应 | 非放射性;样本需求量小 | 需要生物素标记dNTPs;需要磁珠 | [75] | |
| 局部甲基化分析 | 甲基化敏感差异的限制性酶切结合PCR扩增 | 非放射性;最基本的实验硬件条件 | 覆盖有限的CpG位点 | [56] |
| 甲基化敏感差异的限制性酶切结合Souther杂交 | 最基本的实验硬件条件 | 杂交需要放射性标记;覆盖有限的CpG位点 | [42] | |
| 亚硫酸盐测序 | 最基本的实验硬件条件;不依赖于限制性酶切,分辨率达到CpG水平 | 盐硫酸盐处理容易出现一串T,引物不好设计;测序信号不能定量 | [58, 63] | |
| 亚硫酸盐处理结合限制性酶切后PCR扩增(COBRA) | 方法简单,最基本的实验硬件条件; | 引物设计有时候较难;只能覆盖限制性酶切识别的CpG位点 | [61, 62] | |
| 亚硫酸盐处理,亚克隆,测序 | 最基本的实验硬件条件;分辨率达到CpG水平;测序峰图清晰 | 引物设计有时候较难;挑取的克隆多,要求数据统计分析能力强 | [64] | |
| 亚硫酸盐处理,稀释,PCR(数字亚硫酸测序法) | 最基本的实验硬件条件;分辨率达到CpG水平;测序峰图清晰 | 引物设计有时候较难; | [73] | |
| 亚硫酸盐处理,焦磷酸测序 | 辨率达到CpG水平;测序峰图清晰;可以定量 | 扩增片段有限;引物设计有时候较难 | [65] | |
| 亚硫酸盐处理,质谱鉴定 | 不依赖于限制性位点;分析片段比焦磷酸测序大 | 引物设计有时候较难;硬件昂贵;仪器要求非常高级的专业知识 | [66] | |
| COLD-PCR | 非放射性;最基本的实验硬件条件 | 只能覆盖限制性酶切识别的CpG位点 | [70] | |
| 电泳分离 | 非放射性;最基本的实验硬件条件 | 需要变型梯度胶,费时间和人工 | [74] | |
| 生物素-SA-HRP介导的显色反应 | 非放射性;样本需求量小 | 需要生物素标记dNTPs;需要磁珠 | [75] | |
| 基因组尺度的甲基化 | 甲基化敏感差异性酶切结合内部甲基化位点扩增 | 最基本的实验硬件条件;在全基因组尺度检测 | 不是表观遗传组学研究方法,但确实可以检测表观遗传差异;只能覆盖有限的CpG位点 | [76] |
| HELP介导deHpaII小片段富集。富集通过甲基化敏感性差异酶切获得 | 最基本的实验硬件条件;在全基因组尺度检测 | 不是表观遗传组学研究方法,但确实可以检测表观遗传差异;只能覆盖有限的CpG位点 | [57] | |
| 甲基化DNA沉淀结合微阵列芯片技术(MeDIP-Chip) | 在全基因组尺度检测;不依赖限制性位点 | 抗体和微阵列需求的仪器昂贵;数据量依赖于生物信息学知识;分辨率 | [77] | |
| McrBC去除甲基化DNA然后和芯片杂交 | 在全基因组尺度检测;不依赖限制性位点,McrBC可以作用所有的甲基化胞嘧啶,覆盖范围广 | 芯片杂交要求的设备昂贵;数据量依赖于生物信息学知识 | [81] | |
| Infinium HumanMethylation27 Bead人甲基化芯片。亚硫酸盐处理检测目的CpG的C/T SNP | 在全基因组尺度检测;在人中有效检测和生物学时间相关区域的甲基化 | 芯片杂交要求的设备昂贵;芯片限于人的样本;只能覆盖有限的CpG位点;数据量依赖于生物信息学知识 | [82, 83] | |
| 甲基化DNA免疫沉淀并测序(MeDIP-Seq) | 在全基因组尺度检测;分辨率达到CpG水平;不依赖限制性位点,覆盖范围广;样本被富集过,导致测序成本低; | 抗体特异性要求高;测序成本高;数据量依赖于生物信息学知识;对于区分甲基化区域来说,和同等方法相比不够强大 | [91] | |
| 甲基化DNA用甲基结合蛋白MeCP2捕获并测序(MethylCapseq) | 在全基因组尺度检测;分辨率达到CpG水平;不依赖限制性位点,覆盖范围广;样本被富集过,导致测序成本低; | 抗体特异性要求高;测序成本高;数据量依赖于生物信息学知识 | [92] | |
| RRBS(Reduced Representation Bisuphite Sequencing) | 在全基因组尺度检测;分辨率达到CpG水平;不依赖限制性位点;样本被精简过,导致测序成本低; | 测序成本高;数据量依赖于生物信息学知识;对甲基化CpG覆盖程度有限 | [93, 94] | |
| 亚硫酸盐处理并结合深度测序 | 在全基因组尺度检测;分辨率达到CpG水平;不依赖限制性位点;可以直接定量分析CpG位点的甲基化 | 测序成本高;数据量依赖于生物信息学知识 | [97, 98] | |
| 单分子实时测序(SMART) | 在全基因组尺度检测;分辨率达到CpG水平;不依赖限制性位点;可以直接定量分析CpG位点的甲基化;不需要亚硫酸盐处理 | 测序成本高;数据量依赖于生物信息学知识 | [99] | |
| Illumina公司的nfinium HumanMethylation45芯片 和VeraCode甲基化分析平台 | 是Infinium HumanMethylation27的升级版;增加了CpG位点覆盖率 | 杂交相关的仪器贵重;仅限于人基因组;数据量依赖于生物信息学知识 | [85, 96] | |
| DNA甲基化动力学 | 不需要亚硫酸盐处理检测额5fC | 非放射性;基于PCR和NGS | 化学标记效率不高;实验需要高技巧 | [100] |
| 甲基化辅助的亚硫酸盐测序(MAB-seq)和精简的MAB-seq(RRMAB-seq) | 在单碱基分辨率水平检测5fC和5caC | 依赖于DNA甲基化反应;成本高 | [101, 102] | |
| Aba-seq | 在基因组尺度单碱基分辨率水平检测5hmC;兼容单细胞检测 | 基于5hmC葡萄糖基化的效率 | [103, 104] |
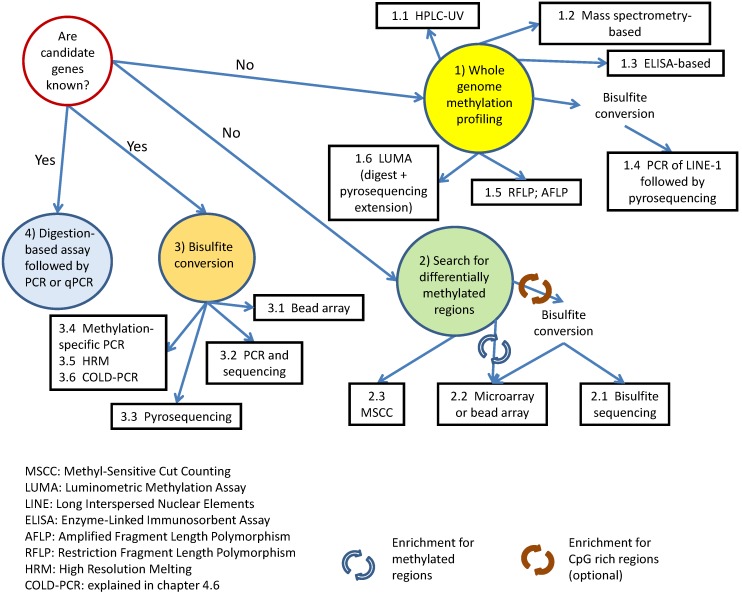
由于表观遗传学研究的进展及表观遗传学变化与几种非传染性疾病的相关性,可靠定量的DNA甲基化谱式分析越来越重要。在过去30年,研究DNA甲基化改变的技术在稳步的发展,其研究方法也从最初忽略局部改变的全局DNA甲基化研究发展到了针对某个特定基因或基因组区域的可靠定量化的甲基化分析,包括目前在全基因组尺度具有CpG分辨率水平解析DNA甲基化变化的研究方法也变得逐渐成熟。特别是近几年随着测序技术的爆发式发展,极大的降低了基于DNA测序技术甲基化研究的成本,这使对DNA甲基化的研究在世界范围内的实验室变得越来越流行。可以预见,在不远的将来随着测序成本的进一步降低,对全基因组所有的CpG位点进行分析会变得可行。然而,针对某个特定研究课题的方法选择仍然是一个不小的挑战。近期发表的一些综述针对性的比较了DNA甲基化研究技术之间的特点 [105] , 诸如预测恰当试验方法的计算机算法等(Fig 8)。
然而,其挑战也是显而易见的。随着数据的大量产生,对其进行生物信息学分析也变得必不可少,特别是对于基因组尺度的研究尤甚。目前表观遗传关注的重点集中于疾病的分子机制和生殖发育生物学。分析方法成本的降低得以使甲基化研究不但在以上这些热门领域里得到广泛的应用,也使之在生物学其他域和更多的模式生物里得以应用。除此之外,成本的降低也促使分析的样本量得到增加,此举产生了更高质量的数据,更利于表观遗传状态的对比。
- Laird P, Jaenisch R. The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet. 1996;30:441-64 pubmed
- Singal R, Ginder G. DNA methylation. Blood. 1999;93:4059-70 pubmed
- Bestor T. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395-402 pubmed
- Jones P, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068-70 pubmed
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261-82 pubmed
- Van den Veyver I. Genetic effects of methylation diets. Annu Rev Nutr. 2002;22:255-82 pubmed
- Yokochi T, Robertson K. Preferential methylation of unmethylated DNA by Mammalian de novo DNA methyltransferase Dnmt3a. J Biol Chem. 2002;277:11735-45 pubmed
- Yoder J, Soman N, Verdine G, Bestor T. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol. 1997;270:385-95 pubmed
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089-93 pubmed
- Edwards T, Myers J. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115:1264-70 pubmed
- Lees-Murdock D, Walsh C. DNA methylation reprogramming in the germ line. Epigenetics. 2008;3:5-13 pubmed
- Allegrucci C, Thurston A, Lucas E, Young L. Epigenetics and the germline. Reproduction. 2005;129:137-49 pubmed
- Durcova-Hills G, Hajkova P, Sullivan S, Barton S, Surani M, McLaren A. Influence of sex chromosome constitution on the genomic imprinting of germ cells. Proc Natl Acad Sci U S A. 2006;103:11184-8 pubmed
- Godfrey K, Lillycrop K, Burdge G, Gluckman P, Hanson M. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R-10R pubmed
- Pogribny I, Ross S, Wise C, Pogribna M, Jones E, Tryndyak V, et al. Irreversible global DNA hypomethylation as a key step in hepatocarcinogenesis induced by dietary methyl deficiency. Mutat Res. 2006;593:80-7 pubmed
- Nan H, Song Y, Yun H, Park J, Kim H. Effects of dietary intake and genetic factors on hypermethylation of the hMLH1 gene promoter in gastric cancer. World J Gastroenterol. 2005;11:3834-41 pubmed
- Lillycrop K, Slater-Jefferies J, Hanson M, Godfrey K, Jackson A, Burdge G. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and . Br J Nutr. 2007;97:1064-73 pubmed
- Champagne F, Weaver I, Diorio J, Dymov S, Szyf M, Meaney M. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. 2006;147:2909-15 pubmed
- Oberlander T, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin A. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97-106 pubmed
- Balaghi M, Wagner C. DNA methylation in folate deficiency: use of CpG methylase. Biochem Biophys Res Commun. 1993;193:1184-90 pubmed
- Soares J, Pinto A, Cunha C, Andre S, Barao I, Sousa J, et al. Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor progression. Cancer. 1999;85:112-8 pubmed
- Goelz S, Vogelstein B, Hamilton S, Feinberg A. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228:187-90 pubmed
- Ben-Hattar J, Jiricny J. Effect of cytosine methylation on the cleavage of oligonucleotide duplexes with restriction endonucleases HpaII and MspI. Nucleic Acids Res. 1988;16:4160 pubmed
- Habib M, Fares F, Bourgeois C, Bella C, Bernardino J, Hernandez-Blazquez F, et al. DNA global hypomethylation in EBV-transformed interphase nuclei. Exp Cell Res. 1999;249:46-53 pubmed
- Friso S, CHOI S, Dolnikowski G, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74:4526-31 pubmed
- Ramsahoye B. Measurement of genome wide DNA methylation by reversed-phase high-performance liquid chromatography. Methods. 2002;27:156-61 pubmed
- Kuo K, McCune R, Gehrke C, Midgett R, Ehrlich M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980;8:4763-76 pubmed
- Song L, James S, Kazim L, Karpf A. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504-10 pubmed
- Kim H, Park J, Jeong K, Lee S. Determining the global DNA methylation status of rat according to the identifier repetitive elements. Electrophoresis. 2007;28:3854-61 pubmed
- Yang A, Estecio M, Doshi K, Kondo Y, Tajara E, Issa J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38 pubmed
- Singer-Sam J, LeBon J, Tanguay R, Riggs A. A quantitative HpaII-PCR assay to measure methylation of DNA from a small number of cells. Nucleic Acids Res. 1990;18:687 pubmed
- Khulan B, Thompson R, Ye K, Fazzari M, Suzuki M, Stasiek E, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046-55 pubmed
- Frommer M, McDonald L, Millar D, Collis C, Watt F, Grigg G, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827-31 pubmed
- Clark S, Harrison J, Paul C, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990-7 pubmed
- Clark S, Statham A, Stirzaker C, Molloy P, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353-64 pubmed
- Eads C, Laird P. Combined bisulfite restriction analysis (COBRA). Methods Mol Biol. 2002;200:71-85 pubmed
- Xiong Z, Laird P. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532-4 pubmed
- Paul C, Clark S. Cytosine methylation: quantitation by automated genomic sequencing and GENESCAN analysis. Biotechniques. 1996;21:126-33 pubmed
- Warnecke P, Stirzaker C, Song J, Grunau C, Melki J, Clark S. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;27:101-7 pubmed
- Tost J, Gut I. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol Biol. 2007;373:89-102 pubmed
- Ehrich M, Nelson M, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102:15785-90 pubmed
- Li L, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427-31 pubmed
- Frigola J, Ribas M, Risques R, Peinado M. Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS). Nucleic Acids Res. 2002;30:e28 pubmed
- Weber M, Davies J, Wittig D, Oakeley E, Haase M, Lam W, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853-62 pubmed
- Tomazou E, Rakyan V, Lefebvre G, Andrews R, Ellis P, Jackson D, et al. Generation of a genomic tiling array of the human Major Histocompatibility Complex (MHC) and its application for DNA methylation analysis. BMC Med Genomics. 2008;1:19 pubmed
- Ordway J, Bedell J, Citek R, Nunberg A, Garrido A, Kendall R, et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis. 2006;27:2409-23 pubmed
- 来邦网
- 英文来邦