
亚细胞分级分离方法概述
An overview of subcellular fractionation methods.
亚细胞分离是研究特定的细胞内结构、细胞器、蛋白或评估这些大分子结构之间联系性的一个重要步骤。亚细胞分级分离利用亚细胞结构的其中一个或多个属性,如浮力密度、表面电荷密度、尺寸和形状等,主要根据其在4℃下高粘度的介质中的差速离心而进行分离。用于差速离心的介质主要有蔗糖、甘露醇,甘油,Ficoll 400(蔗糖的多聚物),Percoll(胶体二氧化硅)和碘克沙醇(OptiPrep)。蔗糖被广泛使用是因为它价格低廉。但这些介质都有各自的优点和局限性, Harford和Bonifacino 在 2011年的文章中对此有详细讨论。本文将主要阐述这些方法,但偏重于那些容易为大多数实验室所操作,而且耗时少的方法,因为快速分离至关重要。凝胶过滤、亲和层析、电泳或选择性密度迁移干扰等方法也可使用。现有操作规程的条件改变是取决于所分离的细胞器、组织或细胞类型和所使用的设备。强烈建议阅读所引用的参考文献中每个操作规程的全部细节。最后,需要通过特定标记物的检测来确定整个分离过程中所搜集的各组分的的纯度和产率。
下面将阐述最常用的亚细胞器分离的方法。鼠肝亚细胞器的分离在文献中已有详细描述而且很容易找见,因此本部分综述的重点是在其他模型系统的亚细胞器分离。
细胞质、核质和染色质组分可以很容易地从收集的培养细胞中制备 [1] 。将细胞重新悬浮在含有0.34 M的蔗糖、10%甘油、低浓度的温和洗涤剂(0.1%的Triton X-100),以及含K +和Mg + 2(保护核不受破裂)的缓冲液中。通过低速离心将细胞核沉淀下来而上清液则为细胞质组分。然后,在含有螯合剂EDTA和EGTA的缓冲液中核进行裂解,不可溶的染色质组分通过低速离心进行沉淀,而上清液则为核质组分 [1] (图1)。
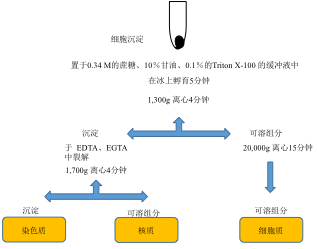
一些研究人员使用一种快速和粗略的方法从细胞质/核质分离制备染色质及其相关蛋白。这种方法简单地通过在含有1%Triton X-100的裂解缓冲液中将细胞进行裂解。在此缓冲液中,染色质和一些细胞骨架结构是不溶性的,它们可以通过离心回收。沉淀物部分可以重新悬浮用于SDS-PAGE电泳之类的 Laemmli样品缓冲液中 [2] 。
- 将单层贴壁或悬浮生长的细胞沉淀物在含有氯化镁和氯化钾的匀浆缓冲液中进行匀浆,然后加蔗糖至浓度为0.25 M,通过低速离心先将核进行沉淀 (1000 g)。把上清液以5000 g进行第二次离心可得到线粒体沉淀。将沉淀物重新悬浮在含蔗糖和Mg+2 的介质中,用Dounce匀浆器进行几次轻柔的匀浆,最后一次离心可以富集线粒体然后将它悬浮在含0.25 M 蔗糖的Tris 缓冲液中或者悬浮在进行下一步分析所需要的缓冲液中(如Laemmli ) [3] 。
- 用酵母裂解酶处理酵母细胞以裂解坚硬的外壁,生成的原生质球用山梨糖醇缓冲液进行洗涤。将所得的沉淀物重新悬浮在含0.6 M的甘露醇匀浆缓冲液,并用Dounce匀浆器进行匀浆几次以裂解。细胞核可以通过低速离心去除,含细胞质的上清液则用一个角度固定的转子以6500 g 速度进行离心而沉淀下线粒体。
此外,也有利用密度梯度分离更为纯化的线粒体的操作规程,但它们耗时更久故而避免使用。虽然差速离心分离线粒体污染有溶酶体和过氧化物酶,但它们是人们所选的方法。因此,所需要的线粒体纯度决定了所需的合适方法。假如 研究新陈代谢,差速离心是首选;假如研究蛋白质的准确定位或者样品纯度是必要条件像蛋白组学研究,那么用密度梯度分离法更为适合。
Smith等 [4] 把经2,000 g 离心的去核上清液再进行20,000 g 离心 30分钟,得到的沉淀含有过氧化物酶体和线粒体,然后将其重新悬浮在MS 缓冲液中( 0.65 M山梨糖醇, 5 mM MES, pH 5.5),加入到同样溶于MS 缓冲液中的Nycodenz 密度梯度的上层 (17%, 25%, 35%, 50%)。 经116,000 g 离心2小时, 过氧化物酶体将会存在于第2 到第8 层中。Fujiki (1982) 和 Nuttley(1990)等进一步 分离了过氧化物酶体膜的相关蛋白,引用参见 [5] 。将前述经20,000 g 离心收集的沉淀重新悬浮在10倍体积的Ti8 缓冲液中 【Tris 10mM, pH 8.0 和PINS (1 mM EDTA, 0.2 mM PMSF, 2 μg leupeptin/ml, 2 μg aprotinin/ml 以及0.4 μg pepstatin A/ml)】, 然后在200,000 g 离心 1 小时。所得的沉淀重新悬浮于Ti8 缓冲液 中并加入0.1 M的碳酸钠再进行 200,000 g 离心1 小时,过氧化物酶体膜就与那些与膜相连但不整合在膜上的蛋白分离开来了。
溶酶体、线粒体和过氧化物酶体在蔗糖梯度中的密度相近,因此最好避免应用此方法分离溶酶体。在Percoll介质中,它们更致密因而可以分离出很少或没有其他细胞器污染的溶酶体。
将洗涤好的细胞悬浮于3毫升含0.25 M的蔗糖缓冲液中,用匀浆器匀浆5次,以800 g 离心10 分钟将完整的核沉淀下来,而碎片和上清液置于冰上。将核沉淀重新悬浮于0.5毫升相同的缓冲液中,同前再次离心并将上清液与第一次离心分离的上清液汇集在一起。在该溶液中加入Percoll原液(含有0.25%蔗糖)至20%的终浓度(注:此浓度可提高到27%-35%),牛血清白蛋白(BSA)至0.4%的终浓度(终体积4.5毫升),于36,000 g 离心30分钟(注:离心条件可为从15,000 g 60分钟至62,500 g 40分钟)。用梯度卸载机将梯度分层收集,每层取0.4毫升,溶酶体通常靠近梯度的底部。为了更好地溶解溶酶体和增加回收率,可以加入NP-40至0.5%的终浓度,然后以100,000g离心1-2小时。但是,如果要研究完整的细胞器例如代谢试验,则应避免用NP-40 [6] 。
Kushimoto和Basrur等人曾用不连续的蔗糖-HEPES缓冲液的梯度来分离黑素体 [7, 8] 。具体地说,将细胞裂解物重新悬浮在2M的蔗糖溶液中,然后加在不连续蔗糖梯度的上层(1.0,1.2,1.4,1.5,1.6,1.8,2.0M)。以100,000g离心1小时后,早期黑色体(I期和II期)大约在1.0-1.2M蔗糖区回收。再将这一富集黑素体的组分在Octopus-PZE 自由流动电泳仪器上用自由流动电泳 (FFE)进一步分离成富含酪氨酸酶和富含蛋白质的组分,速度用2.0毫升/小时。FFE在pH 7.4的0.25M蔗糖三乙醇胺中进行,所用电压为1000-1100 V,电流≈110-125毫安,洗脱流速为3-4毫升/分钟 [8] 。这种方法可以得到高度富集的黑素体样品,用蛋白质组学分析可以鉴定到60 种以上黑素体蛋白 [7] 。
同一组研究人员还创立了另一种可以获取高纯度黑素体组分的方法。将溶于2 M蔗糖中的细胞裂解物铺在不连续蔗糖梯度的底层,经100,000 g 离心1小时,早期的黑素体可在1 M的蔗糖区回收到,然后将此组分铺加在扩展的 0.8, 1.0, 和1.2 M 蔗糖梯度中间并再次离心,这一额外步骤可以去除线粒体污染。后期黑素体(III 期和IV期)也从1.8M的蔗糖层中回收,因为它们含有的黑色素的量较大,因此较重。
Welton等人已经从膀胱癌细胞系HT1376的细胞培养液中纯化出了外来体 [9] 。通过低速简单离心(400 g,10分钟)去除细胞,进而用2000 g 离心15 分钟 和 10,000 g 离心30分钟去除细胞碎片。将上清液铺加在溶于氧化氘(D 2 O或重水)的30%蔗糖垫的上层,高速离心(100,000 g,2小时)。用PBS洗涤一次后,沉淀中包含有外来体。
Hogan等曾用下述方法从尿液中分离外来体 [10] 。将尿液样品先用15,000×g离心清理并用8 μm的尼龙膜过滤,然后再次以150,000 g离心1小时。所得沉淀中含有外来体样的小囊泡和尿中的主要蛋白Tamm-Horsfall蛋白(THP;或尿调节素)。为了去除THP,将样品加在溶于D2O、pH值为6.0的不连续蔗糖梯度(5-30%)上层。以200,000 g离心 24小时后离心后,将样品分14等分收集。将每个组分稀释5至10倍体积的PBS中,然后以150,000 g再次离心1小时,所得的沉淀中含有纯的外来体。
组蛋白是细胞内环境中的最基本的蛋白质。这一特性被用于从细胞或非洲爪蟾提取物中富集组蛋白。将完整、纯化好的核(如先前讨论的)重悬在0.2M硫酸(H2SO4)中,在4℃ 边旋转边温育后,细胞内的大多数蛋白质沉淀下来而组蛋白则仍为可溶,然后经16,000 g离心15分钟可将其回收 [11] 。另一方法是用2.5 M的氯化钠孵育提取染色质组蛋白。
Chen等人曾用下述方法分离高尔基膜 [12] 。将用0.5M蔗糖溶液制备的肝组织匀浆液加在0.86 M蔗糖溶液的上层,然后在上面再加一层0.25 M的蔗糖溶液,于103,800 g离心60分钟后,在0.5-1.3 M的蔗糖溶液层交界处收集膜组分并将其蔗糖浓度调节至0.5M。
中心体可以从贴壁培养的上皮细胞培养中分离但量不大。Andersen等人用多于2x10913] ,将细胞核用低渗裂解液裂解分离,然后经两步离心收集中心体。首先,通过离心分离到50%的蔗糖垫中,随后通过离心分离到40%,50%和70%的蔗糖梯度中。 Moritz等人用如下方法从果蝇胚胎分离中心体:将3.5小时大小的胚胎匀浆液(在BRB80缓冲液+ 100mM的KCl和14%蔗糖中)在1,500 g离心10分钟,除去脂质,上清液用于分离中心体。在上清液中加入终浓度为0.1-0.5%的Triton X-100和50%的蔗糖后,加到蔗糖梯度上(4毫升55%的蔗糖溶液和3毫升70%的蔗糖溶液),在100,000 g 离心90分钟,大多数的中心体积聚在70%缓冲垫的顶部 [14] 。 迁移细胞在一侧上形成一个突起,附着到一个新的点,随后将细胞体的其余部分拉至这一新的附着点。这个突起称为伪足。 Klemke及其同事 [15] 曾运用一种方法将细胞体对趋化剂反应而产生的伪足分离了出来。这一方法通过置于6 孔或24孔培养板的Transwell迁移小室来完成的。简言之,将细胞贴留在具有3微米小孔的滤膜上侧端,因为小孔很小,整个细胞不能通过,但细胞对置于下室中的化学趋化剂反应而形成的伪足则可以通过,它们会附着在滤膜的下侧面上。然后通过固定细胞和伪足,选择裂解液将其裂解收集 [16] 。 Song等人曾从鼠肝脏进行亚细胞分级分离,但此法经过一些改进后也可用于其他组织的亚细胞分级分离 [17] 。图2的示意图总结了整个方法的步骤。简言之,将肝匀浆在1,000 g离心10分钟以分离沉淀(P1)和可溶级分(S1)。 将P1 悬浮在1.8 M 的蔗糖的最终缓冲液中(完整缓冲液 配方读者可参见原文),在70,900 g离心90 分钟 ,得到含有肝细胞核的沉淀 (P2) 和 位于0.25-1.8 M界面之间的可溶组分(S2), P2组分可以储存起来。将S2 重新悬浮在0.25 M的蔗糖溶液中,在1,200 g 离心10分钟得到沉淀含有粗制的质膜,然后将其悬浮于含 1.45 M 蔗糖的最终缓冲液中,在68,400 g 离心 60分钟。 将 位于0.25-1.45 M间的可溶组分加到含0.25 M的蔗糖缓冲液中,以17,600 g再次离心10分钟。 将沉淀重新悬浮在含1.35 M蔗糖的最终缓冲液中,在230,000 g 离心60分钟。回收0.25-1.35 M 间的组分,以0.25 M 蔗糖稀释并在40,000 g再次离心,所得的沉淀含有纯化的质膜蛋白并将其储存。 将可溶性组分S1在8,000 g 重新离心15分钟,所得的不溶成分(P5)含有粗制的线粒体,可溶成分含有内质网(轻的微体和重的微体)和高尔基复合体(S5)。 将P5洗涤后重新悬浮在12毫升含25% Nycodenz [18] 的溶液中并铺加在不连续的Nycodenz梯度(5 毫升 34 %的 Nycodenz和 and 8毫升30 %的Nycodenz)上层,然后再在其上面铺加8毫升23%的 Nycodenz 和 3毫升20%的 Nycodenz。在52,000 g 离心90 分钟后可在25-30% 界面间收集到线粒体组分。 将此组分收集并稀释于最终缓冲液中,此溶液含200 mM 的甘露醇和50 mM蔗糖 。将溶液在15,000 g 离心20分钟,所得的沉淀含有纯的线粒体并将其洗涤并储存。 将 可溶组分S5在34,000 g 离心30 分钟,得到沉淀(P6)和含轻质微体的一个可溶组分(S4)。 将 S4 在 124,000 g 离心从轻质微体分离胞质溶胶并储存。将 P6 与 从前离心所得的轻质微体混合,用含0.25 M蔗糖和0.015 M CsCl 的终溶液稀释,将溶液加在1.3 M的蔗糖溶液上, 在237,000 g 离心2小时,此次离心将粗面内质网(沉淀中)和 光面内质网分离开来,后者位于0.25-1.3 M 的界面。将可溶性组分用0.25 M的蔗糖溶液以1:1 稀释并在124,000 g离心60分钟。 光面内质网可在沉淀中回收到并将其储存 [17] 。 如果未注明引用文献,有关蛋白质功能的信息主要来源于UniprotKB数据库, Cell Signaling和Abcam公司的描述。 将植物组织匀浆重悬在在等渗介质中(0.35 M氯化钠或0.4M蔗糖溶液)以尽量减少对叶绿体的任何破坏,以1000 rpm离心2分钟以除去组织残片和剩余的细胞,然后在3000 rpm下离心5分钟以获得叶绿体沉淀。离心应在0-5°C下进行。 可以。离心力不损伤线粒体结构。此外,离心缓冲液对线粒体有保护作用。 文献18中发表了一个既简单又廉价的用于此分离的操作规程 [19] 。

- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602-12 pubmed
- Attardi G, Ching E. Biogenesis of mitochondrial proteins in HeLa cells. Methods Enzymol. 1979;56:66-79 pubmed
- Smith J, Marelli M, Christmas R, Vizeacoumar F, Dilworth D, Ideker T, et al. Transcriptome profiling to identify genes involved in peroxisome assembly and function. J Cell Biol. 2002;158:259-71 pubmed
- 5.
- Basrur V, Yang F, Kushimoto T, Higashimoto Y, Yasumoto K, Valencia J, et al. Proteomic analysis of early melanosomes: identification of novel melanosomal proteins. J Proteome Res. 2003;2:69-79 pubmed
- Kushimoto T, Basrur V, Valencia J, Matsunaga J, Vieira W, Ferrans V, et al. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc Natl Acad Sci U S A. 2001;98:10698-703 pubmed
- Shechter D, Dormann H, Allis C, Hake S. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445-57 pubmed
- Andersen J, Wilkinson C, Mayor T, Mortensen P, Nigg E, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570-4 pubmed
- Moritz M, Braunfeld M, Fung J, Sedat J, Alberts B, Agard D. Three-dimensional structural characterization of centrosomes from early Drosophila embryos. J Cell Biol. 1995;130:1149-59 pubmed
- Wang Y, Ding S, Wang W, Yang F, Jacobs J, Camp D, et al. Methods for pseudopodia purification and proteomic analysis. Sci STKE. 2007;2007:pl4 pubmed
- Song Y, Hao Y, Sun A, Li T, Li W, Guo L, et al. Sample preparation project for the subcellular proteome of mouse liver. Proteomics. 2006;6:5269-77 pubmed
- Nycodenz® - Density Gradient Media. 来自: www.progen.de/en/nycodenz.html
- 来邦网
- 英文来邦