
The article provides a summary of protein modifications from UniProt database and an overview of research methods for major protein modifications: phosphorylation, acetylation and methylation, ubiquitination, and glycosylation.
人类基因组包含23,000个基因,然而,这些基因产生了显著更大的蛋白质组。这是由于在转录后和翻译后水平的多样性。单个基因在转录后,可以通过可变剪接,偶尔通过RNA编辑,产生多个mRNA分子。许多蛋白质在翻译后,通过蛋白质修饰和/或蛋白质剪切,产生成熟的和功能型的形态。通过广泛的翻译后修饰,蛋白质的结构和功能就多样性了。
在特定的细胞中或在特定的发育阶段,以及在疾病的功能失调状态下,蛋白质修饰的状态和概况揭示了差异蛋白质的功能。这篇综述讨论了蛋白质修饰的四种常见类型及其常用研究方法:磷酸化,乙酰化和甲基化,泛素化和糖基化。
相比于基因组包含的全部基因,人类蛋白质组包含了更多的功能性多肽,部分归因于,翻译的同时和翻译后的蛋白修饰(图1A)。蛋白质组学研究的目的是获得存在于特定的细胞或组织类型,和存在于健康或患病组织的功能蛋白质的全貌。蛋白质组学研究的重要领域之一,是识别那些翻译后修饰的蛋白质,及其修饰位点,确定修饰的功能以及在细胞功能网络中修饰蛋白的相互作用。

在过去的几十年,开发了多种方法测定蛋白质修饰。在这里,我们重点介绍识别蛋白质修饰的一般方法,质谱;识别磷酸化的特异性方法,磷酸盐标记;识别泛素化的方法;在染色质重塑过程中识别组蛋白乙酰化和甲基化的方法;识别蛋白质糖基化的方法(表1和图1B)。详细的实验步骤以后将会整理归纳。
| 修饰 | 功能 | 检测 |
| 磷酸化 | 细胞内信号转导 |
|
| 泛素化 | 蛋白质降解 |
|
| 乙酰化和甲基化 | 转录的调控 |
|
| 糖基化 | 细胞外信号转导 |
|
初步识别新的修饰位点的一个主要方法是计算机分析。已经广泛应用计算机程序根据蛋白质的氨基酸序列识别假定修饰位点,这不是本文的讨论范围。计算机程序的概述见Liu and Li, 2011 [1] 。
在过去20年来,质谱分析已成为确定蛋白质修饰类型和位点的必不可少的工具。质谱分析可用于纯化的蛋白质或蛋白质的混合物,例如细胞裂解液 [2, 3] 。
质谱仪从蛋白样品产生气相离子,根据质荷比(m/z) 将其分开,并记录其丰度。质谱可用于多肽和蛋白质的分子量测定,多肽氨基酸序列的确定,和翻译后修饰的检测,以及多肽和蛋白质的相对定量。这种方法不能用于绝对定量。
质谱分析的样品制备,用限制性内切酶如胰蛋白酶把蛋白质或裂解液消化成小分子多肽片段。然后蒸发和分析这些多肽片段,以确定其m/z值。由于酶切位点是已知的,所以就可以用计算机程序,根据质量确定每个肽段的特定氨基酸序列。由于磷酸基等分子的分子量和电荷是已知的,所以也可以检测肽段中特定氨基酸的磷酸化。
基质辅助激光解吸/电离(MALDI)肽图谱或纳升电质谱仪可以分析酶解消化的蛋白质样品。然而这些方法的局限是不能完全检测所有多肽片段,有些肽段清晰可见,有些肽段不可见。这对于复杂混合物的分析通常是一个主要问题,对于修饰肽段和翻译后修饰的分析,先用反相色谱分离多肽,然后馏分收集和用质谱分析(LC/MS)(图2B) [4] 。为了更准确地确定肽段修饰的性质,经常用串联质谱(MS/MS)实验(图2A)。第一个MS步骤后,用惰性气体撞击多肽离子,从而导致进一步的碎片化。然后在第二个MS步骤分析这些多肽片段 [5] 。在此过程中,有些修饰肽段将保持不变,产生的肽段模式类似于中未相撞的肽段,有些肽段将显著碎片化,产生的肽段模式可以为修饰氨基酸的性质和位点提供进一步的信息。

除了被用来确定单一蛋白质的修饰状态,质谱还被用来确定带有某种特定修饰如磷酸化的所有蛋白质的广泛数据集。这通常涉及到质谱分析前的亲和层析。这种技术被用于确定带有这种修饰的‘次级蛋白质组’,例如磷酸化蛋白质组分析癌症中整个信号网络的激活状态 [6] 。
质谱法已被用来确定上述所有四种修饰。然而,质谱分析较为昂贵,需要特定的设备,通常需要更多的定量数据,所以质谱分析通常是结合其他生化方法用于分析蛋白质的翻译后修饰。多种MS方法的更广泛的综述可参见 [2], [3] 。
磷酸化,或丝氨酸,苏氨酸或酪氨酸残基增加一个磷酸基团,是蛋白质修饰最常见的形式之一(图3A)。在细胞内的信号转导通路,蛋白质磷酸化起着重要的作用,是可逆的,微调的信号 [7] 。几种体内的和体外的方法,被用于检测蛋白质的磷酸化状态,检测一个特定氨基酸的磷酸化,以及确定一个特定的激酶(或磷酸酶)是否作用于目标蛋白质。
放射测量激酶反应使用32P-gamma-ATP,可用于检测体外磷酸化状态 [8] 。这也是检测特定激酶作用的金标准。在反应缓冲液中,温育纯化的靶蛋白,激酶,和32P-gamma-ATP。然后,反应液用滤膜过滤,蛋白质结合到滤膜,洗去未结合的ATP,用闪烁计数器检测磷酸化水平(保留在滤膜的放射性同位素)。或者,反应液用SDS-PAGE电泳分析,用X射线胶片暴光观察。这种方法是半定量,但可以确定磷酸化蛋白的分子量。
同样,反应可以包括磷酸酶,而不是激酶,以确定磷酸化的蛋白是否是一种特定的磷酸酶的底物。
为了检测磷酸化体内,可以使用放射性脉冲标记 [9] 。细胞生长于32P-正磷酸盐存在的条件下。然后用特定抗体免疫沉淀某种蛋白质,沉淀得到的放射性同位素,用闪烁计数器定量或SDS-PAGE电泳分析和X射线胶片暴光。这种方法可以在各种生理条件下检测磷酸化。此外,与蛋白质敲除实验相结合,这种方法也可用来分析一个特定的激酶或磷酸酶是否影响靶蛋白的修饰状态。
磷酸化特异性抗体有两种类别。第一类是通用的 磷酸化酪氨酸,磷酸化丝氨酸,磷酸化苏氨酸抗体,将结合于任何磷酸化酪氨酸,磷酸化丝氨酸和磷酸化苏氨酸分子,与邻近的氨基酸残基无关。第二类包括的抗体是磷酸化特定氨基酸表位抗体 (参见 英文摘要 在来邦网产品数据库的磷酸化特异性抗体)。
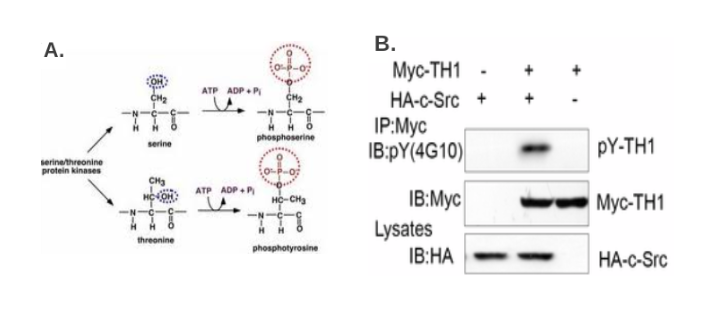
用计算机方法或用质谱法识别了(假定)的磷酸化位点之后,可以从公司购买针对磷酸化位点的通用抗体,通过免疫沉淀的步骤,以确定目标蛋白质是否在特定类型的位点被磷酸化(例如一个典型的cyclin/cdk位点) [10] 。也可以使用针对一个特定的磷酸化氨基酸,如磷酸化酪氨酸,制备的抗体(图3B)。接下来,针对特定表位制备磷酸化抗体,通过简单的免疫印迹检测磷酸化。为了分析大量的样品,可以用酶联免疫吸附试验,目标蛋白结合到膜上,然后用磷酸化特异抗体检测 [11] 。
蛋白质泛素化,或靶蛋白共价结合泛素,在蛋白质降解中的研究最深入。例如,泛素介导的蛋白质降解被证明在细胞周期的运转中发挥了至关重要的作用 [12] 。然而,最近的研究也发现了在多个不关乎蛋白质降解的细胞内信号转导通路中泛素的作用。
泛素介导的信号转导的多样性是取决于目标蛋白可以通过多种方式被泛素修饰 - 在一个或多个位点的单泛素化,和通过几种可能键合的多泛素化 [13] 。每一个泛素分子有七个可能的赖氨酸残基可用于泛素链的延伸; 因此多泛素化的功用取决于泛素链延伸中所用的赖氨酸残基。而这是由特异的E2(泛素结合酶)或E3(泛素连接酶)确定的,但是E1(泛素激活酶)没有特异性(图4A)。特异赖氨酸键合的多泛素链生成了独特的结合表面,与特定相互作用蛋白的泛素结合域相吻合。

泛素化的检测方法可以识别特定赖氨酸残基的泛素化或确认特异的E3(或E2)是否在靶蛋白的修饰上发挥了作用。检测泛素化体内的最直截了当和常用的方法是免疫沉淀认定的泛素化蛋白,然后SDS-PAGE/免疫印迹,观察泛素化蛋白典型的梯状。往往通过异位表达靶蛋白,E3连接酶,甚至泛素,以确定突变的影响 (图4B)。蛋白酶体降解的抑制剂,如MG132,通常被用来分析体内靶蛋白的 K48泛素化(降解锚定)。
体外泛素化检测(提供靶蛋白,泛素,E1,E2,和E3),结合SDS-PAGE,接着蛋白质印迹,可以被用来更直接地检测目标蛋白的泛素化。目标蛋白中赖氨酸残基的突变实验,可以被用来确定泛素的结合位点。相反,在多泛素化的情况下,泛素中特定的赖氨酸残基的突变实验,可以被用来确定特异的泛素键合方式。为了发现新的泛素化蛋白,可以进行泛素介导的蛋白质纯化,然后质谱分析 [14] 。
保守的核心组蛋白H2A,H2B,H3,和H4的表观遗传修饰(如磷酸化,泛素化,乙酰化和甲基化)在基因表达中发挥重要的调节作用 [15] 。例如,组蛋白尾部N-末端赖氨酸残基的乙酰化和甲基化介导染色质域的形成,如,常染色质和异染色质,从而直接介导基因沉默。检测核心组蛋白的修饰状态,是分析基因表达调控的重要技术 [16] 。组蛋白修饰的研究方法的详细讨论 这里.。
所有已知的组蛋白修饰位点的抗体都可以从公司购买。可以使用这些抗体,进行染色质免疫沉淀(ChIP)实验 [17] 。简言之,交联剂如甲醛处理细胞或组织样本,组蛋白交联到染色质。超声或核酸酶处理染色质,产生短的DNA片断。然后用针对组蛋白特定修饰的抗体,进行免疫沉淀(IP)的实验步骤。如果目标基因或启动子中假定的组蛋白修饰位点是已知的,那么PCR就可以对组蛋白某个位点的特定修饰进行定量(图5A)。另外,这种方法可以用来确定通用启动子区域内的组蛋白修饰位点,以及组蛋白某个位点的修饰动力学(图5B)。

如果组蛋白修饰位点是未知的,那么ChIP-on-chip就可以确定基因组中带有特定组蛋白修饰的区域 [18] 。在这个实验中,用不同的荧光素标记IP的DNA片段和IP本身。然后,样品杂交于带有DNA探针的芯片,IP中某个DNA片段的富集与基因组中特定区域的组蛋白特异修饰相关。
ChIP和ChIP-on-chip 实验可以提供某个启动子上组蛋白修饰的信息,修饰状态变化的动力学,以及在某个基因或调控序列中,或在整个基因组中组蛋白修饰所在的位置。这些表观遗传学变化既可以揭示生理功能的正常基因调控,又可以揭示疾病发生的异常基因调控。
在所有的蛋白质中,将近50%的蛋白被糖基化,或结合多种糖基,在多种多样的生物过程中,从胚胎发育,细胞分裂,到蛋白质结构调控,糖基化都发挥作用。在正常的生理活动和疾病(例如炎症,败血症和癌症)过程中,多种不同蛋白质的糖基化状态发生显著变化。因此,检测蛋白质糖基化的实验,有助于疾病预后和治疗目的的研究。
蛋白糖基化的两个主要类型是N-糖基化(聚糖结合到天冬酰胺)和O-糖基化(聚糖结合到丝氨酸或苏氨酸)。不同于以上讨论的蛋白修饰,糖基化修饰的聚糖种类繁多。蛋白糖基化分析的目的是确定聚糖基,被修饰的蛋白质,或结合的位点。在一般情况下,糖基化分析,可以使用三种不同的方法,概述如图6A:化学或酶解后对释放的聚糖本身的分析,胰蛋白酶消化后对糖肽的定性,或完整的糖蛋白中对聚糖的定性。

聚糖分析,可以用质谱分析,有时并用高效液相色谱法。糖蛋白首先经过酶(肽N糖苷酶A或F)或化学(hydazinolysis)方法释放糖基。 然后LC/MS直接分析释放的低聚糖。或者,酶或化学裂解产生可以还原型末端被标上荧光标记,然后通过一些可能的HPLC方法,如亲水作用色谱,分析荧光标记的聚糖 [19] 。
虽然上述方法能提供特定聚糖的信息,但是这些信息不能鉴定出带有特定修饰的蛋白质,也不能鉴定出糖基化的结合位点。为了获得此信息,就要进行蛋白内切酶裂解(如胰蛋白酶消化),LC/MS或HILIC-MS/MS分析糖肽。这种方法提供的信息是带有特定聚糖分子的所有蛋白质,或者是目标蛋白中聚糖的结合位点 [20] 。例如,图6B,是 Cc5细菌培养对特定胎球蛋白残基的糖基化状态的影响,分析方法是胰蛋白酶消化后,用LC-MS检测。
最后,对于有些糖肽,完整蛋白的质谱分析可以提供蛋白质的身份鉴定及其糖基化状态的数据。然而,这种方法取决于糖肽的生化特性,提供的数据的分辨率水平通常低于前述两种方法。
- Mann M, Hendrickson R, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437-73 pubmed
- Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212-7 pubmed
- Korfmacher W. Principles and applications of LC-MS in new drug discovery. Drug Discov Today. 2005;10:1357-67 pubmed
- Verbeck G, Ruotolo B, Sawyer H, Gillig K, Russell D. A fundamental introduction to ion mobility mass spectrometry applied to the analysis of biomolecules. J Biomol Tech. 2002;13:56-61 pubmed
- Lim Y. Mining the tumor phosphoproteome for cancer markers. Clin Cancer Res. 2005;11:3163-9 pubmed
- Cohen P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem Sci. 2000;25:596-601 pubmed
- Hastie C, McLauchlan H, Cohen P. Assay of protein kinases using radiolabeled ATP: a protocol. Nat Protoc. 2006;1:968-71 pubmed
- Peck S. Analysis of protein phosphorylation: methods and strategies for studying kinases and substrates. Plant J. 2006;45:512-22 pubmed
- Zhang H, Zha X, Tan Y, Hornbeck P, Mastrangelo A, Alessi D, et al. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J Biol Chem. 2002;277:39379-87 pubmed
- Blaydes J, Vojtesek B, Bloomberg G, Hupp T. The development and use of phospho-specific antibodies to study protein phosphorylation. Methods Mol Biol. 2000;99:177-89 pubmed
- Tyers M, Jorgensen P. Proteolysis and the cell cycle: with this RING I do thee destroy. Curr Opin Genet Dev. 2000;10:54-64 pubmed
- Tomlinson E, Palaniyappan N, Tooth D, Layfield R. Methods for the purification of ubiquitinated proteins. Proteomics. 2007;7:1016-22 pubmed
- Grant P. A tale of histone modifications. Genome Biol. 2001;2:REVIEWS0003 pubmed
- Spencer V, Sun J, Li L, Davie J. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods. 2003;31:67-75 pubmed
- Buck M, Lieb J. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349-60 pubmed
- Wuhrer M, Deelder A, Hokke C. Protein glycosylation analysis by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;825:124-33 pubmed